Martin L. Pall
Washington State University, 638 NE 41st Avenue, Portland, OR 97232-3312, USA
Received 23 April 2012; Accepted 22 May 2012
Academic Editors: D.-P. Li and B. Waeber
Copyright © 2013 Martin L. Pall. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
The NO/ONOO− cycle is a primarily local biochemical/physiological vicious cycle that appears to cause a series of chronic inflammatory diseases. This paper focuses on whether the cycle causes pulmonary arterial hypertension (PAH) when located in the pulmonary arteries. The cycle involves 12 elements, including superoxide, peroxynitrite (ONOO−), nitric oxide (NO), oxidative stress, NF-κB, inflammatory cytokines, iNOS, mitochondrial dysfunction, intracellular calcium, tetrahydrobiopterin depletion, NMDA activity, and TRP receptor activity. 10 of the 12 are elevated in PAH (NMDA?, NO?) and 11 have documented causal roles in PAH. Each stressor that initiates cases of PAH acts to raise cycle elements, and may, therefore, initiate the cycle in this way. PAH involves a primarily local mechanism as required by the cycle and the symptoms and signs of PAH are generated by elements of the cycle. Endothelin-1, which acts as a causal factor in PAH, acts as part of the cycle; its synthesis is stimulated by cycle elements, and it, in turn, increases each element of the cycle. This extraordinary fit to the principles of the NO/ONOO− cycle allows one to conclude that PAH is a NO/ONOO− cycle disease, and this fit supports the cycle as a major paradigm of chronic inflammatory disease.
1. Introduction
Pulmonary arterial hypertension (PAH) is a progressive and often fatal disease characterized by several important properties including inflammation, oxidative/nitrosative stress, and mitochondrial dysfunction. Such hypertension leads to right ventricular dysfunction that leads in turn to subsequent right heart failure and death. A number of other chronic diseases that share properties described in the first sentence, above, are thought to be caused by a primarily local biochemical vicious cycle, known as the NO/ONOO− cycle (pronounced no, oh no!) [1–11]. Thus the hypothesis being explored in this paper is whether pulmonary hypertension is caused by the local action of the NO/ONOO− cycle in the pulmonary arteries.
One of the testable properties of NO/ONOO− cycle diseases is that for the cycle to be causal, the symptoms and signs of the disease must be generated by elements of the cycle. The classic properties of PAH are vasoconstriction and pulmonary fibrosis and arterial remodeling. Hypertension can be generated by excessive peroxynitrite (ONOO−), which is a vasoconstrictor [12–16], acting in part via oxidative stress and consequent elevated isoprostanes, because isoprostanes are potent vasoconstrictors [12, 13]. Elevated ONOO−can lead to oxidation of tetrahydrobiopterin (BH4), which may lead, in turn, to what is called the partial uncoupling of the nitric oxide synthases (NOSs), leading in turn to chronic ONOO− elevation and, in some cases, chronic hypertension [14–16]. All of these changes, discussed earlier in this paragraph, are thought to be important consequences of the NO/ONOO− cycle and are also thought to be involved in PAH [17, 18]. The properties of peroxynitrite (ONOO−) itself are quite distinct from those of its nitric oxide (NO) precursor, because NO is, of course, a vasodilator.
Pulmonary fibrosis and arterial remodeling are thought to be caused by oxidative stress, inflammatory biochemistry, and mitochondrial dysfunction in the pulmonary arteries [19–22], leading, in turn, to increased hypertension. Because all three of these causes are parts of the NO/ONOO− cycle, the cycle can, in these ways, produce the fibrosis and tissue remodeling that are critical hallmarks of the disease. Consequently both the vasoconstriction and the local fibrosis and remodeling can be understood as being caused, at least in part, by four elements of the NO/ONOO− cycle, elevated ONOO−(previous paragraph), consequent oxidative stress, inflammatory responses and mitochondrial dysfunction.
One of the other testable properties of NO/ONOO− cycle diseases is produced by the primarily local nature of the cycle. Thus, the cycle predicts that if PAH is a NO/ONOO−cycle disease, it will be caused by local action of the cycle in the pulmonary arteries. One type of evidence that strongly supports such a local mechanism is the response of the disease to lung transplantation [23, 24]. The local nature of the fibrosis and remodeling, discussed in the previous paragraph, also supports a local mechanism, as do various other types of histological studies, showing local changes [25–28].
It can be seen from this discussion, that pulmonary hypertension appears to be in good agreement with these two predictions of the NO/ONOO− cycle, suggesting that it may be a NO/ONOO− cycle disease. In order to look at other predictions, we need to consider the properties of the cycle and how well they fit other properties of pulmonary hypertension.
2. Basic Properties of the NO/ONOO− Cycle
The latest version of the NO/ONOO− cycle is diagrammed in Figures 1(a)–1(e)(discussion taken from the author’s web site with permission). Each of the arrows in Figure 1 represents one or more mechanisms by which one element of the cycle can stimulate a second element (see [1–3, 8, 10] for further detailed discussion). Near the core of the cycle (center, slightly left), nitric oxide (NO) reacts with another free radical, superoxide (.O O−) to form ONOO−, a potent nonradical oxidant.
Washington State University, 638 NE 41st Avenue, Portland, OR 97232-3312, USA
Received 23 April 2012; Accepted 22 May 2012
Academic Editors: D.-P. Li and B. Waeber
Copyright © 2013 Martin L. Pall. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
The NO/ONOO− cycle is a primarily local biochemical/physiological vicious cycle that appears to cause a series of chronic inflammatory diseases. This paper focuses on whether the cycle causes pulmonary arterial hypertension (PAH) when located in the pulmonary arteries. The cycle involves 12 elements, including superoxide, peroxynitrite (ONOO−), nitric oxide (NO), oxidative stress, NF-κB, inflammatory cytokines, iNOS, mitochondrial dysfunction, intracellular calcium, tetrahydrobiopterin depletion, NMDA activity, and TRP receptor activity. 10 of the 12 are elevated in PAH (NMDA?, NO?) and 11 have documented causal roles in PAH. Each stressor that initiates cases of PAH acts to raise cycle elements, and may, therefore, initiate the cycle in this way. PAH involves a primarily local mechanism as required by the cycle and the symptoms and signs of PAH are generated by elements of the cycle. Endothelin-1, which acts as a causal factor in PAH, acts as part of the cycle; its synthesis is stimulated by cycle elements, and it, in turn, increases each element of the cycle. This extraordinary fit to the principles of the NO/ONOO− cycle allows one to conclude that PAH is a NO/ONOO− cycle disease, and this fit supports the cycle as a major paradigm of chronic inflammatory disease.
1. Introduction
Pulmonary arterial hypertension (PAH) is a progressive and often fatal disease characterized by several important properties including inflammation, oxidative/nitrosative stress, and mitochondrial dysfunction. Such hypertension leads to right ventricular dysfunction that leads in turn to subsequent right heart failure and death. A number of other chronic diseases that share properties described in the first sentence, above, are thought to be caused by a primarily local biochemical vicious cycle, known as the NO/ONOO− cycle (pronounced no, oh no!) [1–11]. Thus the hypothesis being explored in this paper is whether pulmonary hypertension is caused by the local action of the NO/ONOO− cycle in the pulmonary arteries.
One of the testable properties of NO/ONOO− cycle diseases is that for the cycle to be causal, the symptoms and signs of the disease must be generated by elements of the cycle. The classic properties of PAH are vasoconstriction and pulmonary fibrosis and arterial remodeling. Hypertension can be generated by excessive peroxynitrite (ONOO−), which is a vasoconstrictor [12–16], acting in part via oxidative stress and consequent elevated isoprostanes, because isoprostanes are potent vasoconstrictors [12, 13]. Elevated ONOO−can lead to oxidation of tetrahydrobiopterin (BH4), which may lead, in turn, to what is called the partial uncoupling of the nitric oxide synthases (NOSs), leading in turn to chronic ONOO− elevation and, in some cases, chronic hypertension [14–16]. All of these changes, discussed earlier in this paragraph, are thought to be important consequences of the NO/ONOO− cycle and are also thought to be involved in PAH [17, 18]. The properties of peroxynitrite (ONOO−) itself are quite distinct from those of its nitric oxide (NO) precursor, because NO is, of course, a vasodilator.
Pulmonary fibrosis and arterial remodeling are thought to be caused by oxidative stress, inflammatory biochemistry, and mitochondrial dysfunction in the pulmonary arteries [19–22], leading, in turn, to increased hypertension. Because all three of these causes are parts of the NO/ONOO− cycle, the cycle can, in these ways, produce the fibrosis and tissue remodeling that are critical hallmarks of the disease. Consequently both the vasoconstriction and the local fibrosis and remodeling can be understood as being caused, at least in part, by four elements of the NO/ONOO− cycle, elevated ONOO−(previous paragraph), consequent oxidative stress, inflammatory responses and mitochondrial dysfunction.
One of the other testable properties of NO/ONOO− cycle diseases is produced by the primarily local nature of the cycle. Thus, the cycle predicts that if PAH is a NO/ONOO−cycle disease, it will be caused by local action of the cycle in the pulmonary arteries. One type of evidence that strongly supports such a local mechanism is the response of the disease to lung transplantation [23, 24]. The local nature of the fibrosis and remodeling, discussed in the previous paragraph, also supports a local mechanism, as do various other types of histological studies, showing local changes [25–28].
It can be seen from this discussion, that pulmonary hypertension appears to be in good agreement with these two predictions of the NO/ONOO− cycle, suggesting that it may be a NO/ONOO− cycle disease. In order to look at other predictions, we need to consider the properties of the cycle and how well they fit other properties of pulmonary hypertension.
2. Basic Properties of the NO/ONOO− Cycle
The latest version of the NO/ONOO− cycle is diagrammed in Figures 1(a)–1(e)(discussion taken from the author’s web site with permission). Each of the arrows in Figure 1 represents one or more mechanisms by which one element of the cycle can stimulate a second element (see [1–3, 8, 10] for further detailed discussion). Near the core of the cycle (center, slightly left), nitric oxide (NO) reacts with another free radical, superoxide (.O O−) to form ONOO−, a potent nonradical oxidant.
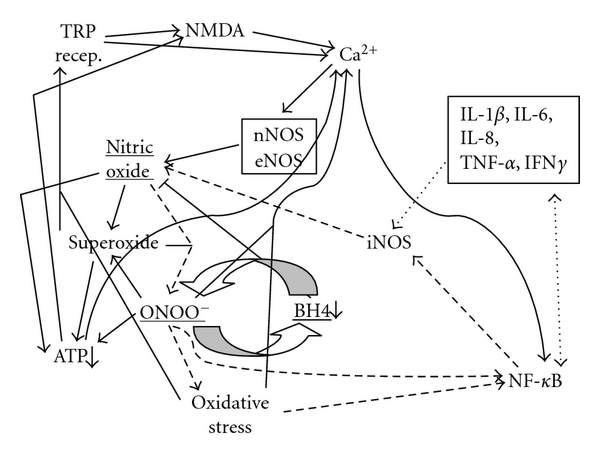
Figure 1: 1(a)–1(e) are essentially identical diagrams of the proposed NO/ONOO− cycle, where each arrow represents one or more mechanisms whereby one element of the cycle acts to increase the levels of a second element of the cycle. Each of these differs from the others in that arrows involved in cycles that constitute parts of the overall NO/ONOO− cycle are dashed, so that these constituent cycles can be considered independently of each other.
Figures 1(a) through 1(e) differ from one another in that each of them diagrams how groups of different mechanisms of the NO/ONOO− cycle forms complete and in most cases multiple cycles which will act to propagate themselves over time, as is the nature of all vicious cycles. Thus without knowing anything about the elements of the cycle, one can see that if these diagrams are correct, each of these parts of the overall cycle (Figures1(a) through 1(e)) will tend to interact with each other through their common elements to form a robust and difficult to downregulate compound cycle that we call the NO/ONOO− cycle. (Note: much documentation for this section is provided in the next section, which focuses on the specific mechanisms of these arrows).
Let us consider dashed arrows in Figure 1(a) starting again from the reaction of NO with superoxide to form peroxynitrite (ONOO−). Elevated ONOO− produces oxidative stress, an imbalance between oxidants and antioxidants. Both ONOO− and oxidative stress activate the transcription factor NF-𝜅B (lower right) which activates, in turn, the transcription of both the inducible nitric oxide synthase gene (iNOS) and also several inflammatory cytokines (box, upper right). Each of these cytokines is linked to NF-𝜅B by a double-headed arrow, such that each of them has its synthesis stimulated by NF-𝜅B and most also, in turn, increase NF-𝜅B activity and some of them can also increase iNOS induction independently of NF-𝜅B. Some of the cytokines can also act independently of NF-𝜅B to increase iNOS activity. Each of these activities, then, can produce increases in iNOS activity, leading, in turn, to increased NO, thus producing a complete cycle.
There are also at least four other major cycles that are each parts of the overall NO/ONOO− cycle. The simplest of these is what is called the central couplet, the reciprocal elevation of ONOO− and depletion of tetrahydrobiopterin (BH4), (slightly below and right of center, Figure 1(c)) [1 𝐺 , 1 𝐼]. ONOO− is known to oxidize and therefore deplete BH4 and BH4 depletion is known to produce a partial uncoupling of the NO synthases (eNOS, nNOS, and iNOS). When these NOSs are uncoupled, they produce superoxide in place of NO. Because the reaction of these two compounds is extremely rapid, but there are mechanisms which lead to rapid loss or sequestration of them in the cell, the synthesis of both on nearby enzymes is expected to be particularly efficient in producing ONOO−, a potent oxidant. Thus, ONOO− will produce BH4 depletion which will be expected to produce more ONOO−. This central couplet is thought to be particularly important in switching on the cycle [8], because NO acts to lower both NF-𝜅B activity and NMDA activity, both important parts of the NO/ONOO−cycle. It can be argued, therefore, that decreasing the ratio of NO to ONOO− may be required to produce a chronic cycle and consequent chronic disease. This central couplet, as discussed below, appears to be particularly important to our understanding of pulmonary hypertension.
Other parts of the cycle (see Figure 1(b)) involve a very complex series of events, both intramitochondrial and also extramitochondrial, leading to mitochondrial dysfunction and consequent ATP depletion (lower, left). The intramitochondrial sequence is often initiated by NO, but involves superoxide, ONOO−, inactivation of mitochondrial proteins, and oxidation of the cardiolipin in the inner membrane in the mitochondrion. The extramitochondrial sequence is triggered by ONOO−, leading to major stimulation of poly (ADP-ribose) polymerase (often designated PARP or PARS), leading to the depletion of the enzyme substrate NAD and consequently also its reduced form, NADH. Depletion of NADH, because it is the most important source of hydrogen reductants entering the mitochondrion, will lead to mitochondrial dysfunction and ATP depletion. Lowered energy metabolism is known to act via two mechanisms to increase activity of the NMDA receptors (Figure 1(b), top center) which acts in turn to increase levels of intracellular calcium and consequent eNOS and nNOS activity (these both being calcium-dependent enzymes), leading to increased NO and ONOO−, feeding back into the mitochondrial cascade and ATP depletion.
An additional cycle (Figure 1(d)) includes three of the TRP group of receptors (upper left) which are known to be stimulated by oxidative stress (TRPA1, TRPV1, and TRPM2); these and other members of this receptor group are also reported to be stimulated by NO. The NMDA receptors, glutamate receptors involved in producing excitotoxicity act, as do the TRP receptors to increase intracellular calcium levels, which act, in turn, to stimulate two of the calcium-dependent NOSs, eNOS, and nNOS, leading back to increased NO, superoxide, ONOO−, and oxidative stress, leading in turn to increased activity of some of these TRP receptors.
Figure 1(e) is focused on the properties of the plasma membrane calcium ATPase, which acts to pump excessive intracellular calcium out of the cell, an enzyme which is inactivated by both ONOO− and other oxidants and being an ATPase, its activity will be, of course, lowered by lowered energy metabolism. All of these interact with each other (Figure 1(e)) to form another complex vicious cycle.
Important, testable predictions of the overall NO/ONOO− cycle are discussed in the second section, below.
3. Specific NO/ONOO− Cycle Mechanisms
What has become known as the NO/ONOO− cycle has become increasingly complex over time, as it has become clear that additional mechanisms should be considered as integral parts of the cycle. The current list of cycle mechanisms is as follows.(1)Extremely rapid diffusion-limited reaction between nitric oxide (NO) with superoxide (OO.−), forming peroxynitrite (ONOO−) [1–3, 5, 29–31].(2)ONOO−, a potent oxidant, can act to increase the activity of the transcription factor NF-𝜅B [5, 32–34].(3)ONOO− breaks down both before and after reaction with carbon dioxide into the following free radicals, hydroxyl (HO), carbonate (CO3), and NO2 radical (NO2), each of which are responsible for a number of consequences produced by ONOO− [1–3, 35, 36].(4)ONOO− being a potent oxidant produces oxidative stress, an imbalance between oxidants and antioxidants [1–3, 30, 31, 35, 36].(5)Oxidative stress also produces increases in NF-𝜅B activity because its activity is stimulated by oxidants and inhibited by chain-breaking antioxidants [2, 32–34,37, 38].(6)NF-𝜅B produces increased transcription of the inducible nitric oxide synthase (iNOS), a gene whose transcription is known to be stimulated by NF-𝜅B elevation [1, 5, 33, 34] and whose elevation also stimulates much of the inflammatory cascade [39].(7)NF-𝜅B also stimulates the transcription of several inflammatory cytokines, including IL-1β, IL-6, IL-8, TNF-𝛼, and IFNγ [1, 5].(8)Each of the cytokines listed in 7 above, act directly and/or indirectly to stimulate the transcription of the iNOS gene, acting in some cytokines via the double-headed arrow linking them to NF-𝜅B and, also, acting in some cytokines directly on iNOS induction [1, 5, 37–42].(9)When iNOS is induced, it produces large amounts of NO.(10)ONOO− inactivates the plasma membrane calcium-ATPase, leading to lowered calcium extrusion and increased levels of intracellular calcium [1, 43].(11)Other oxidants inactivate the plasma membrane calcium-ATPase, leading to increased levels of intracellular calcium [44–48]; such inactivation of the calcium ATPase has substantial pathophysiological effects [45–48] and may well contribute to the prolonged impairment of calcium extrusion seen under circumstances, where the NO/ONOO− cycle may have a role [49–51].(12)Lowered energy metabolism (decreased energy charge/ATP) also lowers calcium-ATPase activity, leading to increased levels of intracellular calcium [52], as predicted for such an ATPase.(13)While modest elevation of mitochondrial calcium, leads to increased ATP synthesis, substantial elevation of intracellular calcium leads to substantial increases in intramitochondrial calcium, leading to increased superoxide generation in the mitochondrion [49–51, 53]; large increases in mitochondrial calcium will lead, in some circumstances, to apoptotic cell death [50, 51, 53].(14)Intracellular calcium stimulates the nNOS and eNOS forms of nitric oxide synthase, both of which are calcium-dependent enzymes.(15)Increased nNOS and eNOS activity both produce increased NO synthesis.(16)ONOO− oxidizes tetrahydrobiopterin (BH4), depleting BH4 levels [1, 2, 8, 10].(17)BH4 depletion produces partial uncoupling of the three NO synthases, such that some of these enzymes produce superoxide in place of NO. Because of the very rapid reaction of these two compounds to produce ONOO−, this partial uncoupling involving nearby NOS enzymes is expected to produce an increase in ONOO− production [8, 10].(18)Nicking of nuclear DNA by ONOO− and hydroxyl and other radicals can produce a massive stimulation of poly ADP-ribose polymerase (PARP) and consequent poly-ADP ribosylation of chromosomal proteins, leading, in turn, to a massive depletion of NAD/NADH pools, because NAD is the substrate for such poly-ADP-ribosylation [1, 2, 54]. NADH depletion lowers, in turn, ATP production in the mitochondrion.(19)Other changes causing ATP depletion come from a cascade of events occurring within the mitochondrion. The cascade starts with NO, possibly produced by mitochondrial NO synthase (mtNOS which is thought to be largely a form of nNOS), with NO binding to cytochrome oxidase, competitively inhibiting the ability of molecular oxygen to bind. This inhibits the ability of cytochrome oxidase to serve as the terminal oxidase of the mitochondrial electron transport chain [1, 2, 55–58].(20)The action of NO, in 19 above, produces increased superoxide production by the electron transport chain [1, 2, 56–58].(21)ONOO− in the mitochondrion also acts to produce increased superoxide from the electron transport chain [1, 2, 56, 58].(22)Peroxynitrite (ONOO−), superoxide, and their products lead to lipid peroxidation of the cardiolipin in the inner membrane of the mitochondrion. Cardiolipin is highly susceptible to such peroxidation, because most of the fatty acids that make up its structure in mammals are polyunsaturated fatty acids, which are much more susceptible to peroxidation than are other fatty acids [1, 2,10, 59–62].(23)Cardiolipin peroxidation leads to lowered activity of some of the enzymes in the electron transport chain, leading to further lowering of ATP synthesis [60–62].(24)Cardiolipin peroxidation also leads to increased superoxide generation from the electron transport chain in the mitochondrion [59, 62].(25)ONOO− produces inactivation of the mitochondrial superoxide dismutase (Mn-SOD) as well as the copper-zinc superoxide dismutase, leading in turn to increased superoxide levels [1, 2, 63–65].(26)ONOO−, superoxide, and NO all inactivate or inhibit the aconitase enzyme, lowering citric acid cycle activity and subsequent ATP synthesis [1, 5, 66].(27)Oxidative stress leads to oxidation of cysteine residues in the enzyme xanthine reductase, converting it into xanthine oxidase which produces superoxide as a product, thus increasing superoxide generation [1, 67].(28)Increased activity of the enzyme NADPH oxidase, which produces superoxide as a product, is an important part of the inflammatory cascade and contributes, therefore, to the cascade by producing increased superoxide [68, 69].(29)Activation of the NMDA receptors allows calcium influx into the cell, raising intracellular calcium levels including mitochondrial calcium levels [1, 2, 9, 49,51, 53].(30)Activity of transfer receptor potential (TRP) receptors also allows calcium influx into the cell, again raising intracellular calcium levels [1, 2], presumably leading to increased nitric oxide production.(31)The main physiological agonist of the NMDA receptors is glutamate whose extracellular concentration is lowered after release by energy-dependent transport. It follows that ATP depletion produces increased NMDA stimulation by lowering glutamate transport [1, 2].(32)The activity of the NMDA receptors is also greatly increased by ATP depletion within the cells containing these receptors. The mechanism here is that the ATP depletion produces partial depolarization of the plasma membrane, which produces, in turn, increased susceptibility of the NMDA receptors to stimulation [1, 2, 9].(33)Three of the TRP group of receptors have been shown to be stimulated by increased superoxide and/or oxidative stress or their downstream consequences, these being the TRPV1, TRPA1, and TRPM2 receptors, with the increased TRPV1, and TRPA1 activity being produced in part through the oxidation of cysteine residue side chains [70–74]. Several TRP receptors are also activated by nitric oxide-mediated nitrosylation [75].(34)TRPV1, TRPA1, and probably several other TRP group receptors, receptor stimulation has each been repeatedly shown to lead to increased NMDA activity [58, 76–89, 91–96], with neurons containing these TRP family of receptors acting in part by releasing glutamate, the major physiological NMDA agonist.
We have, in summary, 34 distinct, well-documented biochemical/physiological mechanisms that make up the complex vicious cycle we call the NO/ONOO− cycle. Most if not all of these are well-accepted biochemistry and physiology and most if not all of these 34 have been shown to play pathophysiological roles in one or more diseases. Consequently, there is little that is new regarding the cycle, except that when the individual mechanisms are put into juxtaposition with each other, they constitute a series of interacting cycles (Figure 1) which, based on their interactions, are likely constitute a robust vicious cycle, the NO/ONOO− cycle, which, is likely to be a major challenge to effectively downregulate.
4. Is Pulmonary Hypertension a NO/ONOO− Cycle Disease? Other Predictions of the Cycle Mechanism
There are five principles that underlie the NO/ONOO− cycle, each of which makes predictions that can be used to determine if a specific disease is a good candidate to be caused by the NO/ONOO− cycle.(1)Short-term stressors that initiate the disease should be able to act by raising cycle elements.(2)The various elements of the cycle, with the possible exception of NO [8], should be elevated in the chronic phase of the disease.(3)The symptoms and signs of the disease should be produced by one or more elements of the cycle.(4)The basic mechanism of the cycle is local and such that it is localized to different tissues in different individuals. The reason for this primarily local nature is that the three inorganic compounds involved, NO, superoxide, and ONOO−, have limited half-lives in biological tissues. And the mechanisms of the cycle, those various arrows, act at the level of individual cells. This allows for great variations in tissue distribution from one patient to another, producing a huge spectrum of illness. The point here is not that there are no systemic changes-clearly antioxidant depletion, neuroendocrine, and immune system changes-and the actions of some inflammatory cytokines will be to some extent systemic. But rather this primarily local nature gives much inherent variation due to the varying tissue localization of the basic mechanism (see Chapter 4, in [1]). A correlate of the primarily local nature of the cycle is that different NO/ONOO− cycle diseases will differ from one another in what tissue or tissues must be impacted by the cycle in order to be diagnosed as a specific cycle-caused disease.(5)Treatment of the disease should involve using agents that lower various parts of the cycle. In other words, we should treat the cause of the disease, not the symptoms.
Evidence has already been provided in the introduction, showing that pulmonary hypertension has a good fit to principles 3 and 4. That is, the symptoms and signs of PAH can be generated by elements of the cycle. In addition, the primarily local nature of the disease has been demonstrated by three different types of observations.
Let us consider the fit to the other three principles.
5. Principle 1
Principle 1 states that if PAH is a NO/ONOO− cycle disease, there should be plausible mechanisms by which stressors that initiate cases of the disease can raise NO/ONOO−cycle elements and thus can, at least in principle, initiate cases of the disease by initiating the cycle. While idiopathic cases of PAH have no identified stressor, other types of cases do have such stressors. In contrast to other proposed NO/ONOO− cycle diseases, it should be note that the stressors implicated in PAH are often chronic stressors rather than short term stressors.
High attitude PAH is a well-established problem in areas of the world where people live at high altitude, notably in regions of central Asia and in the Andes mountains of South America [97–99]. PAH is thought to be triggered, in this condition, by hypoxia. Although animals are susceptible to high altitude PAH [100, 101], most animal model studies of this condition have studied animal responses to hypoxia [102–108].
Khoo et al. [102] studied a mouse model of PAH caused by a mutation that greatly lowers the synthesis of BH4, another cycle element, which causes a greatly increased susceptibility of hypoxia-induced hypertension. A role of BH4 depletion is also suggested by studies showing that Tibetans are genetically resistant to high-altitude PAH and that they also have higher levels of NO exhaled from their lungs. This suggests that Tibetans may have higher levels of BH4, allowing them to synthesize more NO by increasing the coupling of the NOSs to BH4. However, because BH4 levels have not been measured in Tibetans, this interpretation must be viewed as an untested hypothesis.
Other studies implicate NO/ONOO− cycle elements in hypoxic PAH. Fantozzi et al. [103] showed that hypoxia increased calcium influx into human arterial endothelial cells and Remillard and Yuan [99] also implicated increased calcium influx. From these and other studies, Fantozzi et al. [103] conclude that such calcium influx plays a role in “stimulating pulmonary vascular cell proliferation and ultimately, in pulmonary vascular cell remodeling in patients with hypoxia-mediated pulmonary hypertension.” Bartsch et al. [104] showed that the calcium channel blocker nifedipine lowered high-altitude edema associated with high-altitude hypertension, suggesting an important role for elevated intracellular calcium in high-altitude PAH. Wang et al. [105] showed that cell proliferation and intracellular calcium levels were both increased by hypoxia, but that these responses were lowered by capsazepine, a specific antagonist of the TRPV1 receptor. They concluded that “TRPV1 may be a critical pathway or mediator in chronic hypoxia-induced proliferation of human pulmonary artery smooth muscle cells.” Hampl et al. [26] and Palmer et al. [106] showed that hypoxia induces iNOS in the pulmonary arteries. Loot and Fleming [107] showed that hypoxia-mediated vasoconstriction in the mouse was associated with increased calcium influx in mouse pulmonary arteries but that there was much lower calcium influx and much lower vasoconstriction when the arteries came from a transgenic mouse missing the TRPC6 receptor. All three of these studies implicate increased intracellular calcium in PAH and the latter two implicate two members of the TRP receptor family (TRPV1 and TRPC6), still another NO/ONOO−cycle element, in pulmonary hypertension. High-altitude and hypoxia lead to increased activity of the HIF transcription factor which lead, in turn to increased production of endothelin-1 in this condition [108]. As is discussed in the following section of this paper, endothelin-1 elevation leads to increases in most if not all of the NO/ONOO− cycle elements. It can be seen from the above two paragraphs that high-altitude/hypoxic pulmonary hypertension involves elevation of such NO/ONOO− cycle elements as elevated intracellular calcium, TRP receptor activity and iNOS induction; possible BH4 depletion must be viewed as hypothetical. However, other NO/ONOO− cycle elements may be implicated through the elevation of endothelin-1 in high altitude/hypoxic PAH.
Several viral infections, including HIV, HHV-8, and hepatitis B and C, are implicated in producing increased PAH incidence and prevalence [109–114]. It is estimated that HIV infection increases the prevalence of PAH approximately 2500-fold [110].
In the case of HIV, the viral transcriptional factor, Tat has been shown to lead to increased NF-𝜅B activity, increased inflammatory cytokine production, increased 3-nitrotyrosine (a marker for peroxynitrite (ONOO−)), and increased oxidative stress [115]. Thus, all of these elements of the cycle can be produced through the expression of one viral protein. One of these specific cytokines that apparently has a causal role in HIV-associated PAH is IL-6 [116].
While it is clear, from the above, that the HIV-induced increase in PAH incidence and prevalence occurs independently of any antiretroviral drug and that such hypertension can be reduced, in some cases by antiretroviral treatment, there is one antiretroviral drug that has a role in causing PAH. PAH has been shown to be caused by the anti-HIV protease drug ritonavir [117–119]. It apparently does this, at least in part, via increased oxidative stress, because multiple chain-breaking antioxidants greatly lower this effect of the drug [117–119]. Ritonavir has been shown to increase superoxide production [117,120] in mitochondria [121], showing that both superoxide and mitochondrial dysfunction have roles here.
Bacterial infections can also have roles in initiation of cases of PAH. For example, pulmonary tuberculosis often leads to PAH [122–124] and not to, surprisingly, tuberculosis produces substantial elevation of much of the inflammatory cascade, including elevation of NF-𝜅B inflammatory cytokines, iNOS induction, NO, and the marker of peroxynitrite (ONOO−), 3-nitrotyrosine [125].
The role of bacterial infection has been most studied in studies of the action of bacterial endotoxin causing PAH (see, e.g., [126–130]). Endotoxin exposure is known to produce major increases in NF-𝜅B, inflammatory cytokines, iNOS induction leading to increased NO, ONOO−, and oxidative stress, and not surprisingly, these are all found in studies of endotoxin-induced lung injury including PAH [126–130]. Oxidative stress is specifically implicated in having a substantial causal role in initiation of PAH [128]. Furthermore, an important causal role may also be suggested for iNOS induction because of the action of a glucocorticoid in lowering PAH initiation [129]; it is well established that glucocorticoids can lower iNOS induction.
One of the most puzzling issues is the role of liver dysfunction in response to endotoxin exposure clearly shown by the study of Siore et al. [126]. The authors suggest that liver dysfunction may affect this response through such roles as bacterial or endotoxic clearance or roles in producing inflammatory cytokines and eicosanoids [126]. These may be a partial explanation, but there is another explanation for which there appears to be a better precedent. The most important function of the liver is thought to be detoxification of ammonia through the urea cycle and ammonia accumulation is known to be able to produce hepatic encephalopathy. Here ammonia action in the brain, acting via excessive activity of the NMDA receptors, produces the encephalopathy such that the encephalopathy can be greatly lowered by using NMDA antagonists [131–133]. It has been shown that high levels of inhaled ammonia can act to greatly increase lung dysfunction when present along with endotoxin exposure [134]. These considerations raise the question of whether liver dysfunction could cause PAH, in part through ammonia-caused stimulation of NMDA activity in the pulmonary arteries. It is known that there are NMDA receptors in the pulmonary arteries [41, 135, 136], making this interpretation plausible and of course this interpretation provides some support for the view that excessive NMDA activity, an important part of the NO/ONOO− cycle, may have an important role here.
The role of elevated homocysteine in PAH [138–140] also suggests but does not prove a role for the NMDA receptors in PAH, given the known role of homocysteine as an NMDA agonist. A study in pigs showed that a high methionine diet which produces high serum homocysteine produced elevated PAH [141]. The lung dysfunction in this last study [141] was greatly lowered by an angiotensin-converting enzyme inhibitor, strongly suggesting a role for excessive superoxide in homocysteine-initiated PAH as well. This study showing that a simple high methionine diet may cause some cases of PAH suggests a possible initial cause for some cases of what are currently classified as idiopathic PAH and B vitamin/coenzyme deficiencies leading to homocysteine elevation should also be considered as a possible initial cause.
Poisoning by the herbicide paraquat has been shown to cause PAH, with the herbicide acting in an iNOS-dependent manner [142]. Paraquat toxicity also is known to involve mitochondrial dysfunction, excessive NMDA activity, oxidative stress, elevated inflammatory cytokines and elevated NF-𝜅B activity [142–147], raising a possible role for all of these NO/ONOO− cycle elements in the generation of paraquat-dependent PAH.
Systemic autoimmune diseases, including systemic lupus erythematosis, systemic sclerosis and antiphospholipid syndrome are associated with greatly increased incidence of PAH [148–151]. Such autoimmune diseases are well known to increase the inflammatory parts of the cycle, including NF-𝜅B inflammatory cytokine, and iNOS induction with these leading, in turn, to elevate ONOO− and oxidative stress [152]. They may act, therefore, via these mechanisms to turn on the NO/ONOO− cycle.
Inherited cases of PAH occur in PAH families and roughly 3/4 of these are caused by mutations in the BMPR2 gene [153]. Such mutations in this gene, are scattered through much of the gene, such that functions associated with specific parts of the protein encoded by the gene can be ruled out [154]. Because of this, before the recent study of Lane et al. [154], there was no common consequence of the diverse mutations implicated in causing PAH. Lane et al. [154] studied a series of toxic dominant gain of function mutants causing PAH and showed that these all generated increased superoxide-dependent oxidative stress and also mitochondrial dysfunction. They argue that the superoxide-dependent oxidative stress is probably causally involved in PAH and that mitochondrial dysfunction is likely to be causal as well. Thus, three elements of the NO/ONOO− cycle appear to be implicated in this mechanism of disease initiation, namely, increased superoxide, oxidative stress, and mitochondrial dysfunction. It should be noted that genetic initiation involves a very long-term stressor, not a short-term one.
Serotonin (5-HT) and agents that raise serotonin levels and are thought to act via serotonin in case PAH initiation. These agents include fenfluramine, L-tryptophan, cocaine, monocrotaline, and amphetamines, which are all thought to act via elevated serotonin to initiate cases of PAH [155–161]. Oxidative stress responses are involved here [148, 162] as are increased levels of inflammatory cytokines [163]. Serotonin is thought to act, at least in part, by raising levels of a regulatory peptide known as RhoA [155, 156,161]. RhoA is also thought to have roles in several other types of PAH cases including cases involving bleomycin, hypoxia, and endotoxin exposure [162, 164–167] and may, therefore, play a general causal role in PAH. The only way that RhoA can have a causal role in PAH generally, if PAH is a NO/ONOO− cycle disease, is if RhoA acts as part of the cycle.
It is important, therefore, to determine whether RhoA levels may be elevated in response to NO/ONOO− cycle elements and also whether it, in turn, may elevate NO/ONOO−cycle elements. Both of these are predicted if RhoA is acting as part of the NO/ONOO−cycle.
RhoA activity is increased by NF-𝜅B [168, 169] and also by the inflammatory cytokines TNF-𝛼 [169–172] and IL-13 [169, 170]. Both of these cytokines act by raising NF-kappaB when they raise RhoA activity. [168–170]. The inflammatory marker C-reactive protein increased RhoA/Rho-kinase signaling [173]. Jin et al. [174] showed that various free radicals and reactive oxygen species increased RhoA/Rho-kinase signaling. Ryoo et al. [175] showed that oxidative stress, acting through oxidized LDL, stimulated RhoA signaling.
There are a number of studies reporting that RhoA/Rho kinase have roles in generating NO/ONOO− cycle elements, with some of them being done in the context of its role in the vascular epithelia but others done in other pathophysiological contexts. For example, Chandra et al. [176] showed that RhoA/Rho kinase produced increases in ONOO−, superoxide and consequent hydrogen peroxide, and oxidative stress. Resta et al. [177] and also Broughton et al. [178] reported RhoA-dependent apparent increases intracellular calcium levels and also superoxide levels. A Rho kinase inhibitor was shown to decrease both superoxide levels and BH4 depletion and consequent eNOS uncoupling, strongly suggesting that RhoA/Rho kinase raise both superoxide and deplete BH4 [179]. RhoA/Rho kinase increase activity of the inflammatory cytokine/chemokine IL-8 [168].
It can be seen, from the above, that RhoA and RhoA-dependent signaling is both stimulated by three elements of the NO/ONOO− cycle (NF-𝜅B, cytokines, and oxidants/oxidative stress) and that they in turn stimulate multiple elements of the NO/ONOO− cycle, including superoxide, ONOO−, oxidative stress, BH4 depletion, and elevated intracellular calcium, providing support for the view that RhoA functions in these tissues as part of the NO/ONOO− cycle. A similar view was proposed by Yao et al. [180], who in Figure 1 of that paper showed RhoA and the Rho kinase as part of a vicious cycle, with RhoA being stimulated by reactive oxygen species and the cytokine TNF-alpha and RhoA acting in turn, to raise various inflammatory cytokines and other markers, endothelial dysfunction (which is known to involve BH4 depletion) and various oxidants. Elsewhere in that paper [180], they have elevated NF-𝜅B activity as part of their cycle.
The drug bleomycin has been shown to initiate some cases of PAH [162, 181]. It is known to increase several mechanisms involved in the NO/ONOO− cycle including stimulating poly (ADP-ribose) polymerase (PARP), oxidative stress, superoxide generation, inflammatory cytokines, oxidative stress, partial uncoupling of the NOSs (which is presumably caused by BH4 depletion), mitochondrial dysfunction, and NF-𝜅B elevation [181–185]. Superoxide is specifically implicated in having a causal role in bleomycin-initiated PAH because overexpression of superoxide dismutase in a mouse model lessens subsequent pulmonary hypertension, fibrosis, and vascular remodeling following bleomycin treatment [185]. The most directly affected of these is the PARP mechanism because it is greatly stimulated by the single- and double-strand breaks in DNA that are produced by bleomycin. In addition, apoptosis, which is sometimes involved in NO/ONOO− cycle diseases, is also triggered by bleomycin [183]. It can be seen from this, that most of the NO/ONOO− cycle is triggered by bleomycin, such that this alone strongly suggests a NO/ONOO− cycle mechanism for PAH.
An animal model of PAH is caused by a mutation in a gene that produces a deficiency in the production of vasoactive intestinal peptide (VIP) [186, 187]. VIP was also shown to aid in therapy of PAH [188] and a VIP deficiency was shown to produce an inflammatory response [187]. VIP has been shown to stimulate the production of BH4, acting through the synthesis of the enzyme GTP cyclohydrolase I, the rate limiting step in the de novoproduction of BH4 [189]. VIP has also been shown to lower NF-𝜅B activity, inflammatory cytokines and oxidative stress [190, 191]. It follows that a deficiency in VIP will be expected to produce a deficiency in BH4, raise NF-𝜅B activity, inflammatory cytokine levels, and oxidative stress, with all of these being important NO/ONOO− cycle elements.
The studies discussed in the preceding paragraph suggest a causal role for a BH4 deficiency in PAH, but do not prove such a role. However, genetic studies on a BH4-deficient mouse mutant (hph-1), clearly show such a causal role [102, 192, 193]. BH4 deficiency is the well-established cause of nitric oxide synthase uncoupling, leading to increased superoxide production. eNOS uncoupling, a consequence of BH4 depletion, is also found in PAH [194]. BH4 depletion causes lowered eNOS expression (reviewed in [193]), a common correlate found in PAH.
The elements of the cycle implicated in the action various initiators of PAH are summarized in Table 1. It can be seen from Table 1 that each of the elements of the cycle is implicated in action of at least one PAH initiator with most being implicated in multiple initiators. Even PARP, which is often not listed as a NO/ONOO− cycle element but has a major role in one of the two cascades of events leading to mitochondrial dysfunction, is elevated in response to one of the initiators, bleomycin.
Figures 1(a) through 1(e) differ from one another in that each of them diagrams how groups of different mechanisms of the NO/ONOO− cycle forms complete and in most cases multiple cycles which will act to propagate themselves over time, as is the nature of all vicious cycles. Thus without knowing anything about the elements of the cycle, one can see that if these diagrams are correct, each of these parts of the overall cycle (Figures1(a) through 1(e)) will tend to interact with each other through their common elements to form a robust and difficult to downregulate compound cycle that we call the NO/ONOO− cycle. (Note: much documentation for this section is provided in the next section, which focuses on the specific mechanisms of these arrows).
Let us consider dashed arrows in Figure 1(a) starting again from the reaction of NO with superoxide to form peroxynitrite (ONOO−). Elevated ONOO− produces oxidative stress, an imbalance between oxidants and antioxidants. Both ONOO− and oxidative stress activate the transcription factor NF-𝜅B (lower right) which activates, in turn, the transcription of both the inducible nitric oxide synthase gene (iNOS) and also several inflammatory cytokines (box, upper right). Each of these cytokines is linked to NF-𝜅B by a double-headed arrow, such that each of them has its synthesis stimulated by NF-𝜅B and most also, in turn, increase NF-𝜅B activity and some of them can also increase iNOS induction independently of NF-𝜅B. Some of the cytokines can also act independently of NF-𝜅B to increase iNOS activity. Each of these activities, then, can produce increases in iNOS activity, leading, in turn, to increased NO, thus producing a complete cycle.
There are also at least four other major cycles that are each parts of the overall NO/ONOO− cycle. The simplest of these is what is called the central couplet, the reciprocal elevation of ONOO− and depletion of tetrahydrobiopterin (BH4), (slightly below and right of center, Figure 1(c)) [1 𝐺 , 1 𝐼]. ONOO− is known to oxidize and therefore deplete BH4 and BH4 depletion is known to produce a partial uncoupling of the NO synthases (eNOS, nNOS, and iNOS). When these NOSs are uncoupled, they produce superoxide in place of NO. Because the reaction of these two compounds is extremely rapid, but there are mechanisms which lead to rapid loss or sequestration of them in the cell, the synthesis of both on nearby enzymes is expected to be particularly efficient in producing ONOO−, a potent oxidant. Thus, ONOO− will produce BH4 depletion which will be expected to produce more ONOO−. This central couplet is thought to be particularly important in switching on the cycle [8], because NO acts to lower both NF-𝜅B activity and NMDA activity, both important parts of the NO/ONOO−cycle. It can be argued, therefore, that decreasing the ratio of NO to ONOO− may be required to produce a chronic cycle and consequent chronic disease. This central couplet, as discussed below, appears to be particularly important to our understanding of pulmonary hypertension.
Other parts of the cycle (see Figure 1(b)) involve a very complex series of events, both intramitochondrial and also extramitochondrial, leading to mitochondrial dysfunction and consequent ATP depletion (lower, left). The intramitochondrial sequence is often initiated by NO, but involves superoxide, ONOO−, inactivation of mitochondrial proteins, and oxidation of the cardiolipin in the inner membrane in the mitochondrion. The extramitochondrial sequence is triggered by ONOO−, leading to major stimulation of poly (ADP-ribose) polymerase (often designated PARP or PARS), leading to the depletion of the enzyme substrate NAD and consequently also its reduced form, NADH. Depletion of NADH, because it is the most important source of hydrogen reductants entering the mitochondrion, will lead to mitochondrial dysfunction and ATP depletion. Lowered energy metabolism is known to act via two mechanisms to increase activity of the NMDA receptors (Figure 1(b), top center) which acts in turn to increase levels of intracellular calcium and consequent eNOS and nNOS activity (these both being calcium-dependent enzymes), leading to increased NO and ONOO−, feeding back into the mitochondrial cascade and ATP depletion.
An additional cycle (Figure 1(d)) includes three of the TRP group of receptors (upper left) which are known to be stimulated by oxidative stress (TRPA1, TRPV1, and TRPM2); these and other members of this receptor group are also reported to be stimulated by NO. The NMDA receptors, glutamate receptors involved in producing excitotoxicity act, as do the TRP receptors to increase intracellular calcium levels, which act, in turn, to stimulate two of the calcium-dependent NOSs, eNOS, and nNOS, leading back to increased NO, superoxide, ONOO−, and oxidative stress, leading in turn to increased activity of some of these TRP receptors.
Figure 1(e) is focused on the properties of the plasma membrane calcium ATPase, which acts to pump excessive intracellular calcium out of the cell, an enzyme which is inactivated by both ONOO− and other oxidants and being an ATPase, its activity will be, of course, lowered by lowered energy metabolism. All of these interact with each other (Figure 1(e)) to form another complex vicious cycle.
Important, testable predictions of the overall NO/ONOO− cycle are discussed in the second section, below.
3. Specific NO/ONOO− Cycle Mechanisms
What has become known as the NO/ONOO− cycle has become increasingly complex over time, as it has become clear that additional mechanisms should be considered as integral parts of the cycle. The current list of cycle mechanisms is as follows.(1)Extremely rapid diffusion-limited reaction between nitric oxide (NO) with superoxide (OO.−), forming peroxynitrite (ONOO−) [1–3, 5, 29–31].(2)ONOO−, a potent oxidant, can act to increase the activity of the transcription factor NF-𝜅B [5, 32–34].(3)ONOO− breaks down both before and after reaction with carbon dioxide into the following free radicals, hydroxyl (HO), carbonate (CO3), and NO2 radical (NO2), each of which are responsible for a number of consequences produced by ONOO− [1–3, 35, 36].(4)ONOO− being a potent oxidant produces oxidative stress, an imbalance between oxidants and antioxidants [1–3, 30, 31, 35, 36].(5)Oxidative stress also produces increases in NF-𝜅B activity because its activity is stimulated by oxidants and inhibited by chain-breaking antioxidants [2, 32–34,37, 38].(6)NF-𝜅B produces increased transcription of the inducible nitric oxide synthase (iNOS), a gene whose transcription is known to be stimulated by NF-𝜅B elevation [1, 5, 33, 34] and whose elevation also stimulates much of the inflammatory cascade [39].(7)NF-𝜅B also stimulates the transcription of several inflammatory cytokines, including IL-1β, IL-6, IL-8, TNF-𝛼, and IFNγ [1, 5].(8)Each of the cytokines listed in 7 above, act directly and/or indirectly to stimulate the transcription of the iNOS gene, acting in some cytokines via the double-headed arrow linking them to NF-𝜅B and, also, acting in some cytokines directly on iNOS induction [1, 5, 37–42].(9)When iNOS is induced, it produces large amounts of NO.(10)ONOO− inactivates the plasma membrane calcium-ATPase, leading to lowered calcium extrusion and increased levels of intracellular calcium [1, 43].(11)Other oxidants inactivate the plasma membrane calcium-ATPase, leading to increased levels of intracellular calcium [44–48]; such inactivation of the calcium ATPase has substantial pathophysiological effects [45–48] and may well contribute to the prolonged impairment of calcium extrusion seen under circumstances, where the NO/ONOO− cycle may have a role [49–51].(12)Lowered energy metabolism (decreased energy charge/ATP) also lowers calcium-ATPase activity, leading to increased levels of intracellular calcium [52], as predicted for such an ATPase.(13)While modest elevation of mitochondrial calcium, leads to increased ATP synthesis, substantial elevation of intracellular calcium leads to substantial increases in intramitochondrial calcium, leading to increased superoxide generation in the mitochondrion [49–51, 53]; large increases in mitochondrial calcium will lead, in some circumstances, to apoptotic cell death [50, 51, 53].(14)Intracellular calcium stimulates the nNOS and eNOS forms of nitric oxide synthase, both of which are calcium-dependent enzymes.(15)Increased nNOS and eNOS activity both produce increased NO synthesis.(16)ONOO− oxidizes tetrahydrobiopterin (BH4), depleting BH4 levels [1, 2, 8, 10].(17)BH4 depletion produces partial uncoupling of the three NO synthases, such that some of these enzymes produce superoxide in place of NO. Because of the very rapid reaction of these two compounds to produce ONOO−, this partial uncoupling involving nearby NOS enzymes is expected to produce an increase in ONOO− production [8, 10].(18)Nicking of nuclear DNA by ONOO− and hydroxyl and other radicals can produce a massive stimulation of poly ADP-ribose polymerase (PARP) and consequent poly-ADP ribosylation of chromosomal proteins, leading, in turn, to a massive depletion of NAD/NADH pools, because NAD is the substrate for such poly-ADP-ribosylation [1, 2, 54]. NADH depletion lowers, in turn, ATP production in the mitochondrion.(19)Other changes causing ATP depletion come from a cascade of events occurring within the mitochondrion. The cascade starts with NO, possibly produced by mitochondrial NO synthase (mtNOS which is thought to be largely a form of nNOS), with NO binding to cytochrome oxidase, competitively inhibiting the ability of molecular oxygen to bind. This inhibits the ability of cytochrome oxidase to serve as the terminal oxidase of the mitochondrial electron transport chain [1, 2, 55–58].(20)The action of NO, in 19 above, produces increased superoxide production by the electron transport chain [1, 2, 56–58].(21)ONOO− in the mitochondrion also acts to produce increased superoxide from the electron transport chain [1, 2, 56, 58].(22)Peroxynitrite (ONOO−), superoxide, and their products lead to lipid peroxidation of the cardiolipin in the inner membrane of the mitochondrion. Cardiolipin is highly susceptible to such peroxidation, because most of the fatty acids that make up its structure in mammals are polyunsaturated fatty acids, which are much more susceptible to peroxidation than are other fatty acids [1, 2,10, 59–62].(23)Cardiolipin peroxidation leads to lowered activity of some of the enzymes in the electron transport chain, leading to further lowering of ATP synthesis [60–62].(24)Cardiolipin peroxidation also leads to increased superoxide generation from the electron transport chain in the mitochondrion [59, 62].(25)ONOO− produces inactivation of the mitochondrial superoxide dismutase (Mn-SOD) as well as the copper-zinc superoxide dismutase, leading in turn to increased superoxide levels [1, 2, 63–65].(26)ONOO−, superoxide, and NO all inactivate or inhibit the aconitase enzyme, lowering citric acid cycle activity and subsequent ATP synthesis [1, 5, 66].(27)Oxidative stress leads to oxidation of cysteine residues in the enzyme xanthine reductase, converting it into xanthine oxidase which produces superoxide as a product, thus increasing superoxide generation [1, 67].(28)Increased activity of the enzyme NADPH oxidase, which produces superoxide as a product, is an important part of the inflammatory cascade and contributes, therefore, to the cascade by producing increased superoxide [68, 69].(29)Activation of the NMDA receptors allows calcium influx into the cell, raising intracellular calcium levels including mitochondrial calcium levels [1, 2, 9, 49,51, 53].(30)Activity of transfer receptor potential (TRP) receptors also allows calcium influx into the cell, again raising intracellular calcium levels [1, 2], presumably leading to increased nitric oxide production.(31)The main physiological agonist of the NMDA receptors is glutamate whose extracellular concentration is lowered after release by energy-dependent transport. It follows that ATP depletion produces increased NMDA stimulation by lowering glutamate transport [1, 2].(32)The activity of the NMDA receptors is also greatly increased by ATP depletion within the cells containing these receptors. The mechanism here is that the ATP depletion produces partial depolarization of the plasma membrane, which produces, in turn, increased susceptibility of the NMDA receptors to stimulation [1, 2, 9].(33)Three of the TRP group of receptors have been shown to be stimulated by increased superoxide and/or oxidative stress or their downstream consequences, these being the TRPV1, TRPA1, and TRPM2 receptors, with the increased TRPV1, and TRPA1 activity being produced in part through the oxidation of cysteine residue side chains [70–74]. Several TRP receptors are also activated by nitric oxide-mediated nitrosylation [75].(34)TRPV1, TRPA1, and probably several other TRP group receptors, receptor stimulation has each been repeatedly shown to lead to increased NMDA activity [58, 76–89, 91–96], with neurons containing these TRP family of receptors acting in part by releasing glutamate, the major physiological NMDA agonist.
We have, in summary, 34 distinct, well-documented biochemical/physiological mechanisms that make up the complex vicious cycle we call the NO/ONOO− cycle. Most if not all of these are well-accepted biochemistry and physiology and most if not all of these 34 have been shown to play pathophysiological roles in one or more diseases. Consequently, there is little that is new regarding the cycle, except that when the individual mechanisms are put into juxtaposition with each other, they constitute a series of interacting cycles (Figure 1) which, based on their interactions, are likely constitute a robust vicious cycle, the NO/ONOO− cycle, which, is likely to be a major challenge to effectively downregulate.
4. Is Pulmonary Hypertension a NO/ONOO− Cycle Disease? Other Predictions of the Cycle Mechanism
There are five principles that underlie the NO/ONOO− cycle, each of which makes predictions that can be used to determine if a specific disease is a good candidate to be caused by the NO/ONOO− cycle.(1)Short-term stressors that initiate the disease should be able to act by raising cycle elements.(2)The various elements of the cycle, with the possible exception of NO [8], should be elevated in the chronic phase of the disease.(3)The symptoms and signs of the disease should be produced by one or more elements of the cycle.(4)The basic mechanism of the cycle is local and such that it is localized to different tissues in different individuals. The reason for this primarily local nature is that the three inorganic compounds involved, NO, superoxide, and ONOO−, have limited half-lives in biological tissues. And the mechanisms of the cycle, those various arrows, act at the level of individual cells. This allows for great variations in tissue distribution from one patient to another, producing a huge spectrum of illness. The point here is not that there are no systemic changes-clearly antioxidant depletion, neuroendocrine, and immune system changes-and the actions of some inflammatory cytokines will be to some extent systemic. But rather this primarily local nature gives much inherent variation due to the varying tissue localization of the basic mechanism (see Chapter 4, in [1]). A correlate of the primarily local nature of the cycle is that different NO/ONOO− cycle diseases will differ from one another in what tissue or tissues must be impacted by the cycle in order to be diagnosed as a specific cycle-caused disease.(5)Treatment of the disease should involve using agents that lower various parts of the cycle. In other words, we should treat the cause of the disease, not the symptoms.
Evidence has already been provided in the introduction, showing that pulmonary hypertension has a good fit to principles 3 and 4. That is, the symptoms and signs of PAH can be generated by elements of the cycle. In addition, the primarily local nature of the disease has been demonstrated by three different types of observations.
Let us consider the fit to the other three principles.
5. Principle 1
Principle 1 states that if PAH is a NO/ONOO− cycle disease, there should be plausible mechanisms by which stressors that initiate cases of the disease can raise NO/ONOO−cycle elements and thus can, at least in principle, initiate cases of the disease by initiating the cycle. While idiopathic cases of PAH have no identified stressor, other types of cases do have such stressors. In contrast to other proposed NO/ONOO− cycle diseases, it should be note that the stressors implicated in PAH are often chronic stressors rather than short term stressors.
High attitude PAH is a well-established problem in areas of the world where people live at high altitude, notably in regions of central Asia and in the Andes mountains of South America [97–99]. PAH is thought to be triggered, in this condition, by hypoxia. Although animals are susceptible to high altitude PAH [100, 101], most animal model studies of this condition have studied animal responses to hypoxia [102–108].
Khoo et al. [102] studied a mouse model of PAH caused by a mutation that greatly lowers the synthesis of BH4, another cycle element, which causes a greatly increased susceptibility of hypoxia-induced hypertension. A role of BH4 depletion is also suggested by studies showing that Tibetans are genetically resistant to high-altitude PAH and that they also have higher levels of NO exhaled from their lungs. This suggests that Tibetans may have higher levels of BH4, allowing them to synthesize more NO by increasing the coupling of the NOSs to BH4. However, because BH4 levels have not been measured in Tibetans, this interpretation must be viewed as an untested hypothesis.
Other studies implicate NO/ONOO− cycle elements in hypoxic PAH. Fantozzi et al. [103] showed that hypoxia increased calcium influx into human arterial endothelial cells and Remillard and Yuan [99] also implicated increased calcium influx. From these and other studies, Fantozzi et al. [103] conclude that such calcium influx plays a role in “stimulating pulmonary vascular cell proliferation and ultimately, in pulmonary vascular cell remodeling in patients with hypoxia-mediated pulmonary hypertension.” Bartsch et al. [104] showed that the calcium channel blocker nifedipine lowered high-altitude edema associated with high-altitude hypertension, suggesting an important role for elevated intracellular calcium in high-altitude PAH. Wang et al. [105] showed that cell proliferation and intracellular calcium levels were both increased by hypoxia, but that these responses were lowered by capsazepine, a specific antagonist of the TRPV1 receptor. They concluded that “TRPV1 may be a critical pathway or mediator in chronic hypoxia-induced proliferation of human pulmonary artery smooth muscle cells.” Hampl et al. [26] and Palmer et al. [106] showed that hypoxia induces iNOS in the pulmonary arteries. Loot and Fleming [107] showed that hypoxia-mediated vasoconstriction in the mouse was associated with increased calcium influx in mouse pulmonary arteries but that there was much lower calcium influx and much lower vasoconstriction when the arteries came from a transgenic mouse missing the TRPC6 receptor. All three of these studies implicate increased intracellular calcium in PAH and the latter two implicate two members of the TRP receptor family (TRPV1 and TRPC6), still another NO/ONOO−cycle element, in pulmonary hypertension. High-altitude and hypoxia lead to increased activity of the HIF transcription factor which lead, in turn to increased production of endothelin-1 in this condition [108]. As is discussed in the following section of this paper, endothelin-1 elevation leads to increases in most if not all of the NO/ONOO− cycle elements. It can be seen from the above two paragraphs that high-altitude/hypoxic pulmonary hypertension involves elevation of such NO/ONOO− cycle elements as elevated intracellular calcium, TRP receptor activity and iNOS induction; possible BH4 depletion must be viewed as hypothetical. However, other NO/ONOO− cycle elements may be implicated through the elevation of endothelin-1 in high altitude/hypoxic PAH.
Several viral infections, including HIV, HHV-8, and hepatitis B and C, are implicated in producing increased PAH incidence and prevalence [109–114]. It is estimated that HIV infection increases the prevalence of PAH approximately 2500-fold [110].
In the case of HIV, the viral transcriptional factor, Tat has been shown to lead to increased NF-𝜅B activity, increased inflammatory cytokine production, increased 3-nitrotyrosine (a marker for peroxynitrite (ONOO−)), and increased oxidative stress [115]. Thus, all of these elements of the cycle can be produced through the expression of one viral protein. One of these specific cytokines that apparently has a causal role in HIV-associated PAH is IL-6 [116].
While it is clear, from the above, that the HIV-induced increase in PAH incidence and prevalence occurs independently of any antiretroviral drug and that such hypertension can be reduced, in some cases by antiretroviral treatment, there is one antiretroviral drug that has a role in causing PAH. PAH has been shown to be caused by the anti-HIV protease drug ritonavir [117–119]. It apparently does this, at least in part, via increased oxidative stress, because multiple chain-breaking antioxidants greatly lower this effect of the drug [117–119]. Ritonavir has been shown to increase superoxide production [117,120] in mitochondria [121], showing that both superoxide and mitochondrial dysfunction have roles here.
Bacterial infections can also have roles in initiation of cases of PAH. For example, pulmonary tuberculosis often leads to PAH [122–124] and not to, surprisingly, tuberculosis produces substantial elevation of much of the inflammatory cascade, including elevation of NF-𝜅B inflammatory cytokines, iNOS induction, NO, and the marker of peroxynitrite (ONOO−), 3-nitrotyrosine [125].
The role of bacterial infection has been most studied in studies of the action of bacterial endotoxin causing PAH (see, e.g., [126–130]). Endotoxin exposure is known to produce major increases in NF-𝜅B, inflammatory cytokines, iNOS induction leading to increased NO, ONOO−, and oxidative stress, and not surprisingly, these are all found in studies of endotoxin-induced lung injury including PAH [126–130]. Oxidative stress is specifically implicated in having a substantial causal role in initiation of PAH [128]. Furthermore, an important causal role may also be suggested for iNOS induction because of the action of a glucocorticoid in lowering PAH initiation [129]; it is well established that glucocorticoids can lower iNOS induction.
One of the most puzzling issues is the role of liver dysfunction in response to endotoxin exposure clearly shown by the study of Siore et al. [126]. The authors suggest that liver dysfunction may affect this response through such roles as bacterial or endotoxic clearance or roles in producing inflammatory cytokines and eicosanoids [126]. These may be a partial explanation, but there is another explanation for which there appears to be a better precedent. The most important function of the liver is thought to be detoxification of ammonia through the urea cycle and ammonia accumulation is known to be able to produce hepatic encephalopathy. Here ammonia action in the brain, acting via excessive activity of the NMDA receptors, produces the encephalopathy such that the encephalopathy can be greatly lowered by using NMDA antagonists [131–133]. It has been shown that high levels of inhaled ammonia can act to greatly increase lung dysfunction when present along with endotoxin exposure [134]. These considerations raise the question of whether liver dysfunction could cause PAH, in part through ammonia-caused stimulation of NMDA activity in the pulmonary arteries. It is known that there are NMDA receptors in the pulmonary arteries [41, 135, 136], making this interpretation plausible and of course this interpretation provides some support for the view that excessive NMDA activity, an important part of the NO/ONOO− cycle, may have an important role here.
The role of elevated homocysteine in PAH [138–140] also suggests but does not prove a role for the NMDA receptors in PAH, given the known role of homocysteine as an NMDA agonist. A study in pigs showed that a high methionine diet which produces high serum homocysteine produced elevated PAH [141]. The lung dysfunction in this last study [141] was greatly lowered by an angiotensin-converting enzyme inhibitor, strongly suggesting a role for excessive superoxide in homocysteine-initiated PAH as well. This study showing that a simple high methionine diet may cause some cases of PAH suggests a possible initial cause for some cases of what are currently classified as idiopathic PAH and B vitamin/coenzyme deficiencies leading to homocysteine elevation should also be considered as a possible initial cause.
Poisoning by the herbicide paraquat has been shown to cause PAH, with the herbicide acting in an iNOS-dependent manner [142]. Paraquat toxicity also is known to involve mitochondrial dysfunction, excessive NMDA activity, oxidative stress, elevated inflammatory cytokines and elevated NF-𝜅B activity [142–147], raising a possible role for all of these NO/ONOO− cycle elements in the generation of paraquat-dependent PAH.
Systemic autoimmune diseases, including systemic lupus erythematosis, systemic sclerosis and antiphospholipid syndrome are associated with greatly increased incidence of PAH [148–151]. Such autoimmune diseases are well known to increase the inflammatory parts of the cycle, including NF-𝜅B inflammatory cytokine, and iNOS induction with these leading, in turn, to elevate ONOO− and oxidative stress [152]. They may act, therefore, via these mechanisms to turn on the NO/ONOO− cycle.
Inherited cases of PAH occur in PAH families and roughly 3/4 of these are caused by mutations in the BMPR2 gene [153]. Such mutations in this gene, are scattered through much of the gene, such that functions associated with specific parts of the protein encoded by the gene can be ruled out [154]. Because of this, before the recent study of Lane et al. [154], there was no common consequence of the diverse mutations implicated in causing PAH. Lane et al. [154] studied a series of toxic dominant gain of function mutants causing PAH and showed that these all generated increased superoxide-dependent oxidative stress and also mitochondrial dysfunction. They argue that the superoxide-dependent oxidative stress is probably causally involved in PAH and that mitochondrial dysfunction is likely to be causal as well. Thus, three elements of the NO/ONOO− cycle appear to be implicated in this mechanism of disease initiation, namely, increased superoxide, oxidative stress, and mitochondrial dysfunction. It should be noted that genetic initiation involves a very long-term stressor, not a short-term one.
Serotonin (5-HT) and agents that raise serotonin levels and are thought to act via serotonin in case PAH initiation. These agents include fenfluramine, L-tryptophan, cocaine, monocrotaline, and amphetamines, which are all thought to act via elevated serotonin to initiate cases of PAH [155–161]. Oxidative stress responses are involved here [148, 162] as are increased levels of inflammatory cytokines [163]. Serotonin is thought to act, at least in part, by raising levels of a regulatory peptide known as RhoA [155, 156,161]. RhoA is also thought to have roles in several other types of PAH cases including cases involving bleomycin, hypoxia, and endotoxin exposure [162, 164–167] and may, therefore, play a general causal role in PAH. The only way that RhoA can have a causal role in PAH generally, if PAH is a NO/ONOO− cycle disease, is if RhoA acts as part of the cycle.
It is important, therefore, to determine whether RhoA levels may be elevated in response to NO/ONOO− cycle elements and also whether it, in turn, may elevate NO/ONOO−cycle elements. Both of these are predicted if RhoA is acting as part of the NO/ONOO−cycle.
RhoA activity is increased by NF-𝜅B [168, 169] and also by the inflammatory cytokines TNF-𝛼 [169–172] and IL-13 [169, 170]. Both of these cytokines act by raising NF-kappaB when they raise RhoA activity. [168–170]. The inflammatory marker C-reactive protein increased RhoA/Rho-kinase signaling [173]. Jin et al. [174] showed that various free radicals and reactive oxygen species increased RhoA/Rho-kinase signaling. Ryoo et al. [175] showed that oxidative stress, acting through oxidized LDL, stimulated RhoA signaling.
There are a number of studies reporting that RhoA/Rho kinase have roles in generating NO/ONOO− cycle elements, with some of them being done in the context of its role in the vascular epithelia but others done in other pathophysiological contexts. For example, Chandra et al. [176] showed that RhoA/Rho kinase produced increases in ONOO−, superoxide and consequent hydrogen peroxide, and oxidative stress. Resta et al. [177] and also Broughton et al. [178] reported RhoA-dependent apparent increases intracellular calcium levels and also superoxide levels. A Rho kinase inhibitor was shown to decrease both superoxide levels and BH4 depletion and consequent eNOS uncoupling, strongly suggesting that RhoA/Rho kinase raise both superoxide and deplete BH4 [179]. RhoA/Rho kinase increase activity of the inflammatory cytokine/chemokine IL-8 [168].
It can be seen, from the above, that RhoA and RhoA-dependent signaling is both stimulated by three elements of the NO/ONOO− cycle (NF-𝜅B, cytokines, and oxidants/oxidative stress) and that they in turn stimulate multiple elements of the NO/ONOO− cycle, including superoxide, ONOO−, oxidative stress, BH4 depletion, and elevated intracellular calcium, providing support for the view that RhoA functions in these tissues as part of the NO/ONOO− cycle. A similar view was proposed by Yao et al. [180], who in Figure 1 of that paper showed RhoA and the Rho kinase as part of a vicious cycle, with RhoA being stimulated by reactive oxygen species and the cytokine TNF-alpha and RhoA acting in turn, to raise various inflammatory cytokines and other markers, endothelial dysfunction (which is known to involve BH4 depletion) and various oxidants. Elsewhere in that paper [180], they have elevated NF-𝜅B activity as part of their cycle.
The drug bleomycin has been shown to initiate some cases of PAH [162, 181]. It is known to increase several mechanisms involved in the NO/ONOO− cycle including stimulating poly (ADP-ribose) polymerase (PARP), oxidative stress, superoxide generation, inflammatory cytokines, oxidative stress, partial uncoupling of the NOSs (which is presumably caused by BH4 depletion), mitochondrial dysfunction, and NF-𝜅B elevation [181–185]. Superoxide is specifically implicated in having a causal role in bleomycin-initiated PAH because overexpression of superoxide dismutase in a mouse model lessens subsequent pulmonary hypertension, fibrosis, and vascular remodeling following bleomycin treatment [185]. The most directly affected of these is the PARP mechanism because it is greatly stimulated by the single- and double-strand breaks in DNA that are produced by bleomycin. In addition, apoptosis, which is sometimes involved in NO/ONOO− cycle diseases, is also triggered by bleomycin [183]. It can be seen from this, that most of the NO/ONOO− cycle is triggered by bleomycin, such that this alone strongly suggests a NO/ONOO− cycle mechanism for PAH.
An animal model of PAH is caused by a mutation in a gene that produces a deficiency in the production of vasoactive intestinal peptide (VIP) [186, 187]. VIP was also shown to aid in therapy of PAH [188] and a VIP deficiency was shown to produce an inflammatory response [187]. VIP has been shown to stimulate the production of BH4, acting through the synthesis of the enzyme GTP cyclohydrolase I, the rate limiting step in the de novoproduction of BH4 [189]. VIP has also been shown to lower NF-𝜅B activity, inflammatory cytokines and oxidative stress [190, 191]. It follows that a deficiency in VIP will be expected to produce a deficiency in BH4, raise NF-𝜅B activity, inflammatory cytokine levels, and oxidative stress, with all of these being important NO/ONOO− cycle elements.
The studies discussed in the preceding paragraph suggest a causal role for a BH4 deficiency in PAH, but do not prove such a role. However, genetic studies on a BH4-deficient mouse mutant (hph-1), clearly show such a causal role [102, 192, 193]. BH4 deficiency is the well-established cause of nitric oxide synthase uncoupling, leading to increased superoxide production. eNOS uncoupling, a consequence of BH4 depletion, is also found in PAH [194]. BH4 depletion causes lowered eNOS expression (reviewed in [193]), a common correlate found in PAH.
The elements of the cycle implicated in the action various initiators of PAH are summarized in Table 1. It can be seen from Table 1 that each of the elements of the cycle is implicated in action of at least one PAH initiator with most being implicated in multiple initiators. Even PARP, which is often not listed as a NO/ONOO− cycle element but has a major role in one of the two cascades of events leading to mitochondrial dysfunction, is elevated in response to one of the initiators, bleomycin.

Table 1: NO/ONOO− cycle elements implicated in PAH initiation.
While each of the cycle elements is implicated in the action of initiators of PAH, that does not necessarily mean that they all have important causal roles in PAH. It is possible that some may be epiphenomena, occurring but not playing any causal role. It is important, therefore, to look at which elements have causal roles in the initiation process, based either on genetic studies or on the use of specific inhibitors, or both. The studies cited above in this section provide evidence for causal roles in PAH initiation for BH4 depletion, superoxide elevation, oxidative stress, TRPV1 & TRPC6 activity, and the cytokine IL-6.
6. Endothelin-1 as an Important Causal Factor in PAH
Endothelin-1 (ET-1) is a potent vasoconstrictor 21-amino acid residue protein, generated by proteolysis of a larger precursor protein [103A]. It has an important role in regulating vascular tone [195, 196], and its levels have been shown to be substantially elevated in PAH [197]. Furthermore, an ET-1 receptor antagonist is thought to be an effective treatment for PAH [198]. These observations and many others that have confirmed them argue strongly that ET-1 has an important causal role in PAH. And this, in turn, provides both an important challenge and important test of the NO/ONOO− cycle mechanism for PAH.
The only way that the NO/ONOO− cycle can be causal for PAH if ET-1 elevation is also causal is for ET-1 elevation to be part of the cycle in PAH. That is, the NO/ONOO− cycle, if it applies to PAH, predicts that ET-1 levels must be elevated in response to one or more cycle elements and must act, in turn, to raise various cycle elements. These are both strong predictions of the NO/ONOO− cycle mechanism, if it applies to PAH, strong predictions that are not likely to be made based on any other unrelated hypothesis. Are they both correct? The following studies involve the ET-1 role in PAH but also in other tissues.
ET-1 has been shown to be induced by inflammatory cytokines [199–201]. It has also been shown to be induced by oxidants with such induction also lowered by antioxidants [202–204]. The activity of ET-1 has been shown to be increased by both NF-𝜅B action and also the action of another inflammatory cytokine activated pathway [205–207]. Thus, the inflammatory part of the NO/ONOO− cycle has a powerful role in increasing ET-1 activity. Furthermore BH4 was shown to substantially lower ET-1 levels [208], so that BH4 depletion, another part of the NO/ONOO− cycle has a role in raising ET-1 levels. ET-1 levels are also reported to be increased by TRPV1 [209, 210], another part of the cycle. It follows from these observations that elements of the NO/ONOO− cycle (at least five, in this case) have substantial roles in raising ET-1 levels, as predicted if PAH is a NO/ONOO− cycle disease and that ET-1 elevation also has a causal role in PAH.
Furthermore, ET-1 action has been shown to elevate, in turn, essentially the entire NO/ONOO− cycle. It increases oxidative stress [211–217], superoxide [211, 213–216], the ONOO− marker 3-nitrotyrosine [211, 212], NF-𝜅B activity [211, 217], and NO [211, 213,218], lowers levels of BH4 [213–215, 217, 219], stimulates iNOS induction [211, 217], increases TRP receptor function [220, 221], increases levels of intracellular calcium [222–224], increases NMDA activity [225, 226], produces mitochondrial dysfunction [227, 228], and increases inflammatory cytokine levels [206, 207, 217]. All of these provide evidence for elevation of each of the NO/ONOO− cycle elements.
However, there is one fact that requires some explanation. Even though there are studies showing that ET-1 increases iNOS induction [211, 217], there are also studies showing that it can decrease iNOS induction [219, 229]. These latter studies are in situations where substantial iNOS was already induced before ET-1 exposure and where lowered iNOS induction was accompanied by BH4 depletion [219, 229]. It has been shown that high level iNOS induction requires substantial pools of BH4 [230]. It is the author’s view, therefore, that this lowered iNOS induction is probably caused by the BH4 depletion produced by ET-1 and therefore that these observations do not conflict with the conclusion that ET-1 acts by elevating the NO/ONOO− cycle. ET-1 lowers induction of the enzyme GTP cyclohydrolase I, the rate limiting step in the production of BH4 [216], providing further evidence for a BH4 role here.
In conclusion, then, there is substantial evidence that elevated ET-1 can be induced by five elements of the NO/ONOO− cycle and that it can act, in turn, to elevate all of the NO/ONOO− cycle elements. It follows that ET-1 acts, at least in part, by elevating the NO/ONOO− cycle in PAH.
7. Elevated NO/ONOO− Cycle Elements in the Chronic Phase of PAH and Therapy via Agents that Lower NO/ONOO− Cycle Elements
The second principle of the NO/ONOO− cycle is that the elements of the cycle should be elevated in the chronic phase of the disease. And the fifth principle is that NO/ONOO−cycle diseases should respond to therapies that lower NO/ONOO− cycle elements and should, therefore, be treated in this way. The studies supporting both of these principles in PAH often deal with both of them, so both of these principles will be treated together here. We have already discussed substantial evidence for this in PAH in the context of the action of the various stressors that initiate PAH—evidence discussed in the previous two sections of this paper. Many of the studies cited in those two sections are not only relevant to the issue of disease initiation but also are relevant to the issue of the chronic phase of the disease. This section considers still additional studies that relate to these same issues, are elements of the cycle elevated in the chronic phase of PAH? Are agents that lower NO/ONOO− cycle elements helpful in therapy. Much of this discussion focuses on animal model studies.
Bowers et al. [28] was a histological study of human lung tissue in PAH, which showed that several markers of oxidative stress were elevated, that 3-nitrotyrosine, a marker of ONOO− was elevated and that inflammatory markers were elevated as well. They suggest that [28] chronic prostacyclin infusion, an anti-inflammatory treatment is helpful, suggesting that agents lowering the inflammatory part of the cycle may be useful in therapy.
Lakshminrusimha et al. [231] found that isoprostane, a marker of oxidative stress, and 3-nitrotyrosine, a marker of ONOO−, were both elevated in PAH and that addition of intratracheal superoxide dismutase (SOD) an enzyme that degrades superoxide, produced symptomatic improvement and lowered both of these markers. Interestingly, they also showed that inhaled NO produced symptomatic improvement but also raised ONOO−.
Herget et al. [232] reported increased NO production and elevated 3-nitrotyrosine in hypoxic PAH and also showed that antioxidants were helpful in prevention of PAH. This study, implicates NO, ONOO−, and oxidative stress and suggests that antioxidants may be helpful in therapy.
Weissmann et al. [233] reviewed evidence that both superoxide and its product, hydrogen peroxide, are elevated in PAH and argue that both mitochondrial and extramitochondrial mechanisms, including NADPH oxidase, are involved in superoxide generation. They also argue for a role of oxidant damage in causing the tissue remodeling in PAH.
Van Rheen et al. [185] studied normal mice and those expressing much higher levels of extracellular superoxide dismutase (SOD) in PAH initiated by bleomycin. The vascular remodeling and PAH were greatly lowered by SOD and survival was greatly improved. These studies provide strong support for the view that superoxide has an important role in tissue remodeling and PAH and that an agent that acts to lower superoxide is, therefore very helpful in prevention.
Dorfmüller et al. [234] showed that a marker of oxidative stress, malondialdehyde, 3-nitrotyrosine (ONOO− marker), and an inflammatory cytokine were all elevated in the tissue remodeling in PAH. There were also changes in expression of antioxidant enzymes, providing further support for a role of oxidative stress.
Rashid et al. [235] showed that a superoxide dismutase mimic greatly lowered the development of PAH, suggesting a causal role for superoxide and showing that an agent that lowers superoxide was very useful in prevention.
Lu et al. [236] showed that the drug, rosiglitazone, a PPAR𝛾 agonist, has favorable properties on human pulmonary artery smooth muscle, acting to lower two responses that are both elevated in response to hypoxia: increased NF-𝜅B activity and increased expression of one of the NADPH oxidase genes, Nox4. Rosiglitazone also acts to lower PAH responses. Because NADPH oxidase produced superoxide, this also implicates superoxide in PAH. These studies show that an agent that lowers both superoxide production and NF-𝜅B activity is helpful in therapy.
Oxidative stress and reactive oxygen species were discussed as causal factors in generating the symptoms and signs of PAH in the introduction to this paper [17, 22].
7.1. Peroxynitrite (ONOO−)
There are many papers, showing the 3-nitrotyrosine, a marker for ONOO− elevation, is raised in PAH, including four that were discussed above in this section [28, 185, 231,232]. In another study, Oishi et al. [237], showed that both superoxide and ONOO− were elevated following rebound from NO inhalation. So there should be no question that ONOO− is elevated in PAH. What is the main focus, here, therefore, is the study of acausal role of ONOO− in PAH.
Agbani et al. [238] showed that ONOO− stimulated the growth of pulmonary artery smooth muscle cells, a response involved in the remodeling response in PAH; the growth response was blocked by ebselen, an ONOO− scavenger, providing further evidence for a causal role for ONOO−.
Aggarwal et al. [239] showed that addition of an NO donor to pulmonary artery endothelial and smooth muscle cells increased ONOO− and increased nitration of protein kinase G. This response was shown to be blocked by either superoxide dismutase (SOD) or by an SOD mimic drug. This study followed an earlier one showing that nitration of the G kinase lowered its responsiveness to cGMP stimulation and therefore lowered its ability to produce NO-stimulated vasodilation and such attenuated vasodilation was also demonstrated in this study [239]. One part of the study showed that inhaled NO produced elevation of ONOO−, increased G kinase nitration, and decreased G kinase activity, providing a mechanism for lowered responsiveness to inhaled NO with time. Interestingly, in one part of the study [239], L-arginine was found to lower both ONOO− following simultaneous NO inhalation and also helped to maintain G kinase activity; this is important because L-arginine increases NO synthesis while lowering superoxide production from uncoupled nitric oxide synthases; the arginine finding argues, therefore, that superoxide generated on the uncoupled nitric oxide synthase enzyme may have a key role here. The causal role of ONOO− here is demonstrated by its known role in protein nitration, the demonstrated causation of lowered G kinase activity by a compound that breaks down to produce ONOO− and also the demonstrated causal role in this study of both of the ONOO− precursors, NO, and superoxide. This is a clear study demonstrating that an important change in the vasculature in PAH, namely, lowered responsiveness to NO mediated vasodilation, is caused by ONOO− mediated nitration of a particular protein, the G kinase. It also shows the efficacy of agents lowering superoxide.
In another study, Agbani et al. [240] demonstrated that ONOO− stimulates pulmonary endothelial cell and smooth muscle proliferation by a regulatory pathway involving both ERK and PKC protein kinases as well as two growth factor receptors. The causal role of ONOO− was demonstrated here, both by adding pure peroxynitrite (ONOO−) and by using the ONOO− scavenger ebselen to block these responses [240]. Belik et al. [241] asked whether hypoxia-caused PAH and vascular remodeling were caused by ONOO−elevation by using a ONOO− decomposition catalyst, FeTPPS. They showed that FeTPPS blocked some but not other changes [241], showing that those that were blocked were probably produced by ONOO−. Masood et al. [242] used this same FeTPPS decomposition catalyst to determine whether ONOO− was responsible for various changes in the pulmonary vasculature produced by 60% oxygen exposure in neonatal rats, leading to PAH. They concluded that some but not other changes were caused by ONOO−. It may be inferred from the above studies that many of the changes seen with PAH in the pulmonary vasculature are caused by ONOO−, the most central element in the NO/ONOO− cycle but that there are others that are not caused by ONOO− but perhaps may involve other cycle elements.
7.2. NF-𝜅B
One study discussed earlier in this section, Lu et al. [236] implicated elevated NF-𝜅B activity in PAH as did several studies cited in the previous two sections of the paper. There are several additional studies that implicate elevated NF-𝜅B activity in PAH. In one of these, Liu et al. [243] studied increased NF-𝜅B expression that developed along with PAH. The statin drug simvastatin was shown to have a favorable effect, apparently in part at least, by suppressing the increase in NF-𝜅B expression. A study by Li et al. [244] was not a study on PAH but was a study of the effect of an inflammatory protein found in PAH, C-reactive protein on the expression of NF-𝜅B and also the inflammatory cytokine IL-6 in pulmonary artery smooth muscle cells. They showed that both NF-𝜅B expression and IL-6 expression were raised by C-reactive protein and that these raises were suppressed by another statin drug, atorvastatin. Both of these studies provide some support for the view that increased NF-𝜅B expression and in the latter study, the cytokine IL-6, may have roles in PAH and that suppressing these roles with statin drugs may be useful therapeutically.
Another study suggesting a causal role of NF-𝜅B elevation is that of Kimura et al. [245], which showed that a nanoparticle-mediated delivery of an NF-𝜅B decoy (i.e., a nucleic acid-like sequence to which NF-𝜅B binds) to the lungs, greatly attenuated inflammation and smooth muscle proliferation in a monocrotaline PAH model. They also reported NF-𝜅B activity elevation in the lungs of patients with PAH.
Sawada et al. [246], studying a monocrotaline rat model of PAH, found prolonged elevation of NF-𝜅B activity. They used the NF-𝜅B inhibitor pyrrolidine dithiocarbamate and showed that it lowered NF-𝜅B activity and ameliorated the PAH development. Their results both confirm a role of NF-𝜅B elevation in PAH and suggest that agents lowering NF-𝜅B activity should be useful in therapy.
Huang et al. [247] did a similar study, also in a monocrotaline rat PAH model but got very different results. They found only a brief elevation of NF-𝜅B activity following monocrotaline treatment and did not confirm the lowering of NF-𝜅B activity by pyrrolidine dithiocarbamate. They state that the “reason for the discrepancy between our results and those of Sawada et al. is not clear.”
Two additional studies showed that NF-𝜅B activity can produce vascular smooth muscle proliferation [248, 249]. In one of these studies [248], antisense polynucleotides blocked expression of one of the protein subunits of NF-𝜅B and blocked vascular smooth muscle proliferation. In the other study [249], a hyperactive mutant gene for the inhibitor of NF-𝜅B IkappaBalpha, greatly lowered NF-𝜅B activity and also blocked vascular smooth muscle proliferation. These studies show that vascular smooth muscle proliferation can be caused by NF-𝜅B activity elevation and also lowered by agents that lower NF-𝜅B activity.
In summary, there is substantial evidence that elevated NF-𝜅B activity has a role in PAH and that it has a causal role in the development of PAH, being specifically implicated in smooth muscle proliferation and therefore in tissue remodeling.
7.3. Inflammatory Cytokines
There have been numerous studies reporting elevated levels of inflammatory cytokines in PAH, including two discussed above in this section [234, 243]. A number of such studies reported elevation of multiple inflammatory cytokines/chemokines in PAH. For example, Soon et al. [250] reported that IL-1𝛽, −2, −4, −6, −8, −10, and −12p70, as well as TNF-𝛼were elevated in PAH patients. Elevated levels of four of these were associated with lowered survival, with IL-6 showing the highest such association. Li et al. [251] found that 11 inflammatory proteins were elevated in adventitial fibroblasts from calves suffering from hypoxic PAH, most of these proteins different from the ones studied by Soon et al. [250], but including IL-1𝛽 and IL-6. Yu et al. [252] reported elevated levels of IL-1𝛽, TNF-𝛼, and IL-6 in PAH patients undergoing hemodialysis. Sin and Man [253] also provided evidence for elevation of IL-1𝛽, TNF-𝛼, and IL-6 in PAH patients. Hamal et al. [254] showed that PAH induced by injection of microparticulate cellulose into susceptible chickens was accompanied by elevated expression of several inflammatory cytokines including IL-1𝛽, IL-4, IL-6, IL-8, and IFN𝛾. An IL-1 receptor antagonist was shown to lower PAH responses, suggesting a causal role for IL-1𝛽 elevation in PAH [255]. Wanderer provided a strong rationale for using IL-1 𝛽 antagonists or receptor blockers in PAH treatment [256].
A number of studies have focused, to some extent, specifically on a role for IL-6 in PAH. Kalambokis et al. [257] reported high serum levels of IL-6 in cirrhosis-associated PAH. Chaouat et al. [258] found that COPD patients with PAH had higher levels of IL-6 than did COPD patients without PAH. They also showed that individuals carrying a genetic polymorphism in the IL-6 gene associated with higher gene expression had significantly higher prevalence of PAH, strongly suggesting a causal role for IL-6. Finally they showed [258] that pulmonary artery smooth muscle cells exposed to hypoxic conditions had twice the IL-6 mRNA as those not exposed to hypoxic conditions. The concluded that “inflammation, most likely involving IL-6, may contribute substantially to PH complicating COPD.” Furuya et al. [259] reviewed evidence implicating IL-6 in promoting smooth muscle and epithelial proliferation in PAH. They showed that a patient with severe refractory PAH responded dramatically to treatment with a humanized monoclonal antibody to the IL-6 receptor and suggest that IL-6 blockade may be a promising adjunct treatment for PAH. Steiner et al. [260] compared transgenic mice overexpressing IL-6 with normal mice. They found that such transgenic mice differed from normals under both hypoxic and normoxic conditions, leading to increased occurrence of a number of changes characteristic of PAH. They suggest that “IL-6 promotes the development and progression of pulmonary vascular remodeling and PAH through proproliferative and antiapoptotic mechanisms.”
It may be seen from the above two paragraphs, that there are wide ranging proinflammatory changes associated with PAH and that IL-6 elevation may play a particularly important role in PAH development. One study [259] suggests that IL-6 action may be an important therapeutic target in PAH and two other studies [255, 256] suggest that IL-1𝛽 action may be as well.
7.4. iNOS Induction
iNOS induction has been reported in two additional studies to be implicated in PAH [261, 262]. In one of these [262], a specific inhibitor of iNOS was shown to greatly lower PAH development, arguing for a causal role of iNOS induction in PAH. An iNOS knockout mouse was less sensitive to exacerbation of PAH, including inflammatory responses, as discussed above [263].
7.5. Mitochondrial Dysfunction
Quite a number of studies have reported substantial mitochondrial dysfunction in PAH [264–269]. The genetic studies in [266–268] strongly suggest that such mitochondrial dysfunction can have a strong, causal role in PAH and one study used mitochondrial inhibitors which showed a complex causal role in PAH [269]. Four of these studies [264,266, 268, 269] all suggest mechanisms by which mitochondrial dysfunction may contribute to the symptoms and signs of PAH. However, none of these provide any evidence that agents that improve mitochondrial function can prevent or produce improvements in PAH. This last issue of possible therapy via improved mitochondrial seems to be a neglected one in PAH. The only such studies that the author is familiar with are two studies showing that inhibitors of poly (ADP-ribose) polymerase (or synthetase) (PARS or PARP) provide substantial protection from PAH [270, 271]. This PARP activity is known to initiate one of the two cascades of events, leading to mitochondrial dysfunction, proposed to be part of the NO/ONOO− cycle, and therefore lowering its activity will be predicted to have a major effect in lowering mitochondrial dysfunction, as has been shown in a number of chronic inflammatory diseases.
7.6. TRP Receptors and Also Intracellular Calcium
The study of Yu et al. [272] showed that a single nucleotide polymorphism in the TRPC6 gene, introducing an NF-𝜅B site in the enhancer region of the gene, increased susceptibility to PAH. This study implicates both a TRP receptor and consequent increased intracellular calcium in PAH. Two studies, in addition to the ones cited above, also suggest roles of the TRP group of receptors and increased intracellular calcium in PAH [273, 274]. Capsaicin is a TRPV1 agonist which over the longer term, produces very substantial down-regulation of the TRPV1 and several other TRP receptors. It is interesting, therefore that several studies showed that capsaicin pretreatment over a period of days produces greatly decreased PAH induction [275–277].
There are a variety of additional studies showing that elevated intracellular calcium levels have causal roles in PAH [273–282], including studies using calcium channel blockers [279–282] to obtain clinical improvement. It follows that agents lowering intracellular calcium may also be useful in therapy. A role for intracellular calcium is further supported by a study showing that inhibition of calcium-dependent protease, calpain blocked the development of several features of PAH [283].
7.7. NO Elevation?
The NO/ONOO− cycle mechanism predicts that NO synthesis will be increased through iNOS induction and by calcium-stimulation of eNOS and nNOS activity but that it will also be decreased through BH4 depletion and consequent partial uncoupling of all three NOSs [8]. Furthermore, as discussed above, BH4 depletion can lead to lowered eNOS and iNOS expression. Consequently, it is unclear whether the cycle mechanism should predict whether an increase or decrease in NO should be expected. However, most proposed NO/ONOO− cycle diseases have published studies showing apparent increases in NO synthesis.
It is important to raise the question as to whether NO should be considered an element of the cycle or not. Clearly, the answer is yes, if one considers that NO is essential for the production of peroxynitrite (ONOO−), but the answer is less clear as to whether its synthesis must be elevated in NO/ONOO− cycle diseases. These considerations should be kept in mind in considering the data on NO in PAH.
Most studies of NO in PAH patients have measured exhaled NO levels from the lungs in order to assess possible changes in NO synthesis in PAH [284–290]. NO is relatively stable in the gas phase, allowing for such measurements, whereas the instability of NO in biological fluids means that estimates of NO synthesis from blood measurements are usually done via nitrate/nitrite products derived from NO. However the instability of NO in biological fluids means that anything that lowers the movement of NO from the pulmonary arteries into the gas phase, such as fibrosis, arterial remodeling, or lowered ciliary activity may be expected to lower measured exhaled NO. Furthermore, the reaction of NO with superoxide to form ONOO− (both superoxide and ONOO− are clearly both elevated in PAH), will also lower any measurement of exhaled NO.
The majority of studies on exhaled NO in PAH patients report lowered levels when compared with controls [284–287, 289], but two such studies report apparent elevated levels, with one reporting a nonsignificant trend [288] and the other significant elevation [290]. Consequently, there appears to be substantial variation in these studies. Two of these studies compared exhaled NO levels with nitrate/nitrite levels in the blood in the same individuals. Both found that although exhaled NO levels were lower in PAH patients, blood levels of nitrite/nitrate were elevated [284, 289]. The interpretations of these results by the authors of these two studies were different from one another, but clearly one interpretation is the one suggested in the previous paragraph, that NO synthesis may be elevated but that movement of NO from the pulmonary arteries to the gas phase may be substantially slowed in PAH. If this interpretation is correct, it raises questions about whether any of the studies of lowered exhaled NO in PAH should be interpreted as good measurements of NO synthesis. In addition, three studies reported that therapies that produced symptomatic improvement in PAH patients, lowered exhaled NO levels [285, 291, 292], suggesting a correlative relationship between lowered NO and improved symptoms.
The observations discussed in the previous two paragraphs show that NO is not likely to be consistently low in PAH and that correlative data suggest that therapies lowering NO can produce therapeutic improvement, although none of these therapies were designed to lower NO. Before leaving this issue, it is important to discuss an additional type of observation on PAH. It has often been reported that lowered eNOS expression is found in the pulmonary arteries and associated cells in PAH, and such observations have often been used to suggest that low NO helps cause PAH. However, it should be noted that BH4 depletion, a part of the cycle that has been confirmed in PAH, causes lowered expression of eNOS (reviewed in [193]). It is possible, therefore, that such lowered eNOS expression is a protective mechanism, aimed at avoiding excessive superoxide production, rather than a causal mechanism in PAH; if this view is correct, then it follows that this protective mechanism is insufficient to avoid PAH under conditions where the disease is induced.
In conclusion, then, none of the studies on NO in PAH can be argued to conflict with the NO/ONOO− cycle mechanism, although like many other types of data, there are multiple interpretations of these data that can be suggested.
One of the questions that must be raised in the context of a possible NO/ONOO− cycle mechanism for PAH is what role NO has in the process? Clearly inhaled NO can produce an initial lowering of hypertension, something that might be expected, based on the role of NO as a vasodilator and also possibly the action of NO in lowering two NO/ONOO−cycle elements, NF-𝜅B activity and NMDA activity. However, the most crucial NO role in the cycle is acting as a precursor for ONOO− and consequently a pathophysiological role for NO is an essential part of the cycle. Such a pathophysiological role for NO in PAH is most clearly seen in the rebound that occurs after NO inhalation, when NO is no longer producing favorable responses and therefore is no longer inhaled. The rebound response to previous NO inhalation clearly shows that NO can produce a chronic exacerbation of the disease and clearly establishes such a pathophysiological role. However, a question should be raised is whether the properties of that chronic exacerbation are consistent with that predicted by the NO/ONOO− cycle There are a number of studies that suggest that they are. For example, Weinberger et al. [293] showed that previous NO exposure led not only to elevated ONOO−, as predicted by the NO precursor role in ONOO−formation, but also increased superoxide production and iNOS activity, consequences that must be indirect but are predicted by the cycle. Wedgwood et al. [212] showed a role for both increased ONOO− and superoxide in the rebound response following NO inhalation. Shanley et al. [263] showed inflammatory responses following NO inhalation, responses that were greatly lowered in an iNOS knockout mouse, showing that such inflammatory responses were not only produced by such NO inhalation, but were lowered by blocking an important part of the NO/ONOO− cycle, namely iNOS induction. The Lakshminrusimha et al. study [231] showed that inhaled NO produced increases in ONOO−. The Aggarwal et al. study [239] showed that inhaled NO not only raised ONOO−, but also was ONOO− dependent nitration of the G kinase enzyme, leading to lowered vasodilation, an important part of the rebound phenomenon. Oishi et al. [237] found that both ONOO− and superoxide were elevated following NO inhalation and established a causal role of superoxide in the rebound response by using polyethylene glycol-conjugated SOD to inhibit the rebound response.
All of these findings are consistent with upregulation of the NO/ONOO− cycle as a consequence of inhaled NO, with NO acting as a ONOO− precursor. The knockout iNOS mouse study suggests that lowering NO can be helpful and can lower the inflammatory parts of the cycle.
This does not mean that there are no situations where NO can be helpful, certainly its role in vasodilation and its potential role in lowering both NMDA receptor activity and NF-𝜅B activity can also be helpful, but in a NO/ONOO− cycle situation, its role as a precursor of ONOO− must be considered to be paramount.
7.8. BH4 Therapy
Several studies implicating BH4 depletion in PAH were discussed in the previous two sections of this paper. Three studies suggest that agents that raise BH4 levels may be helpful in therapy. One was a study on the safety of sapropterin (the drug name for BH4) in PAH patients [294].
In another paper, Teng et al. [295] studied the pulmonary artery endothelial tissues from fetal lambs with pulmonary hypertension comparing these is normal lambs. In this study Teng et al. [295] showed that the BH4 levels in the PAH lambs were very low compared with normals, leading to eNOS uncoupling, lowered NO and raised superoxide. Interestingly, the lowered BH4 levels were not only produced by ONOO− oxidation of BH4, but also by lowered GTP cyclohydrolase I, the rate limiting enzyme in BH4 synthesis. Addition of sepiapterin, a BH4 precursor to these PAH arterial tissues, raised BH4, raised NO synthesis and lowered superoxide. It also led to increased probable stability of the eNOS enzyme by increased association with Hsp90 protein. Most importantly it help restore angiogenesis which is deficient in PAH. The authors suggest that raising BH4 levels is important in PAH therapy.
A third study showed that heat treatment, such as that found in sauna therapy, was therapeutically useful in PAH patients [296]. Sauna therapy is thought to act by raising the level of the rate limiting enzyme in BH4 synthesis, GTP cyclohydrolase I, and therefore raising BH4 availability [297].
While none of these studies can be taken as definitive, they suggest that raising BH4 levels is likely to be useful in PAH therapy.
7.9. NMDA Activity?
Evidence suggesting excessive activity of the NMDA receptors in PHA, discussed above such a mechanism in case initiation by endotoxin exposure, elevated homocysteine or paraquat exposure, exacerbation by ammonia, or the evidence, also discussed above, showing that ET-1 raised NMDA activity. Each of these observations are susceptible to various interpretations, however. Similarly, while there have been no studies aimed at testing the efficacy of agents lowering NMDA activity in PAH, five studies have shown the value of raising magnesium levels in PAH [298–303]. The NMDA receptors are highly susceptible to being activated in conditions of even marginal magnesium deficiency, so an attractive interpretation of the magnesium studies is that magnesium is acting by lowering NMDA activity. Other interpretations are possible here, as well. Clearly, this is the part of the cycle that is most weakly linked to PAH and it may be argued that it may not have a causal role at all.
8. Is Resveratrol a Magic Bullet for the Treatment of PAH?
Some very exciting research has shown that an agent that gives very good clinical responses in preventing PAH acts to down-regulate most of the NO/ONOO− cycle elements. This research was performed by Csiszar, Ungvari, and their colleagues at New York Medical College. In one study [304], they showed the following NO/ONOO− cycle elements were elevated in PAH: oxidative stress, the inflammatory cytokines IL-6 and TNF-𝛼, other inflammatory genes, NF-𝜅B and iNOS induction. All of these were blocked along with the PAH by resveratrol, the stilbene flavonoid reported to be a wide ranging health-promoting agent. In another study [305] resveratrol prevented PAH development along with elevation of three cytokines, oxidative stress, endothelial dysfunction, and upregulation of NADPH oxidase, one of the mechanisms that generates superoxide. In a third study [306], this one on coronary arterial endothelial cells, resveratrol improved mitochondrial function, increased the mitochondrial SOD (an enzyme that gets rid of superoxide), increased reduced glutathione (an important antioxidant mechanism), lowered oxidative stress, lowered mitochondrial production of reactive oxygen species, all favorable changes lowering NO/ONOO− cycle elements. A study by another research group [307] showed that resveratrol increased mitochondrial biogenesis and lowered angiotensin-II-dependent activity, thus providing improvements via two distinct mechanisms. Another group [308] showed that resveratrol increases synthesis of all three superoxide dismutases (not just the mitochondrial enzyme), lowered NADPH oxidase (an important source of superoxide), lowered superoxide (not surprisingly), lowered oxidative stress, and lowered ONOO−. Most importantly, they also showed that the synthesis of BH4 was increased by inducing increases levels of the enzyme GTP cyclohydrolase I, the rate limiting enzyme in the de novo pathway for BH4 synthesis. Not surprisingly, the increased BH4 led to increased coupling of the eNOS enzyme [308]. Other studies have shown that resveratrol decreases the activity of the NMDA receptors and also the related kainate receptors, acting on both the receptor activities themselves [309–311] and also acting to increase glutamate transport [312], thus lowering the extracellular glutamate that acts as an NMDA agonist. In each of these actions, resveratrol is thought to act by stimulating the activity of SIRT1, a protein that has a wide ranging activity in regulating gene expression. All of this suggests that resveratrol is almost a dream agent for lowering the NO/ONOO− cycle. By raising SIRT1, it lowers, at least in some tissues each of the following NO/ONOO− cycle elements: oxidative stress, ONOO−, mitochondrial dysfunction, superoxide, inflammatory cytokines and other inflammatory markers, NF-𝜅B and excessive NMDA activity while raising BH4 levels. Almost the whole cycle is lowered by resveratrol.
Should we be surprised that another research group asked [313]: is resveratrol the magic bullet for pulmonary hypertension?
It is this author’s view that resveratrol is likely to be a useful therapeutic agent for PAH but that it is not likely to be a magic bullet. The basis of this assessment is two-fold. Firstly, it is much more difficult, in general, to cure disease than to prevent it and this is to be especially expected for such a mechanism as the NO/ONOO− cycle, whose robust structure (Figures 1(a)–1(e)) strongly argues that it is difficult to down-regulate. These studies were basically preventative rather than curative [304–306]. Furthermore, the activity of SIRT1 [314], which mediates each of these resveratrol effects, is sensitive to oxidative inactivation and is also dependent on NAD but NAD can be massively depleted by the elevation of PARP activity by ONOO– in the NO/ONOO– cycle (see number 18 of the 34 mechanisms). It follows from this that repleting NAD with high doses of nicotinamide and nicotinic acid (forms of niacin) will be needed as well as the use of antioxidants and lowering of ONOO−, for resveratrol to be very active. In general, the more severe the disease and the cycle, the more difficult it will be to do all that effectively.
9. Discussion and Conclusions
The NO/ONOO− cycle is a complex biochemical/physiological vicious cycle that can explain various chronic inflammatory diseases localized to certain regions of the body. It should be considered as an etiologic mechanism only for such inflammatory diseases that can be initiated by stressors, including short-term stressors, that can elevate cycle elements and can, therefore, at least in principle, initiate the cycle through such elevation. Clearly PAH is such a disease and, therefore, PAH may be a candidate for such an etiology. What should be clear from this paper, is that there are many diverse studies on PAH, each implicating NO/ONOO− cycle elements in PAH (much of these are summarized in Table 2). While many of these do not show that these elements have causal roles in causing PAH, so that they may simply be an epiphenenon simply based on those studies, there are also many studies that do show a causal role for 11 out of 12 cycle elements. These collectively therefore provide strong support for a NO/ONOO− cycle etiology.
While each of the cycle elements is implicated in the action of initiators of PAH, that does not necessarily mean that they all have important causal roles in PAH. It is possible that some may be epiphenomena, occurring but not playing any causal role. It is important, therefore, to look at which elements have causal roles in the initiation process, based either on genetic studies or on the use of specific inhibitors, or both. The studies cited above in this section provide evidence for causal roles in PAH initiation for BH4 depletion, superoxide elevation, oxidative stress, TRPV1 & TRPC6 activity, and the cytokine IL-6.
6. Endothelin-1 as an Important Causal Factor in PAH
Endothelin-1 (ET-1) is a potent vasoconstrictor 21-amino acid residue protein, generated by proteolysis of a larger precursor protein [103A]. It has an important role in regulating vascular tone [195, 196], and its levels have been shown to be substantially elevated in PAH [197]. Furthermore, an ET-1 receptor antagonist is thought to be an effective treatment for PAH [198]. These observations and many others that have confirmed them argue strongly that ET-1 has an important causal role in PAH. And this, in turn, provides both an important challenge and important test of the NO/ONOO− cycle mechanism for PAH.
The only way that the NO/ONOO− cycle can be causal for PAH if ET-1 elevation is also causal is for ET-1 elevation to be part of the cycle in PAH. That is, the NO/ONOO− cycle, if it applies to PAH, predicts that ET-1 levels must be elevated in response to one or more cycle elements and must act, in turn, to raise various cycle elements. These are both strong predictions of the NO/ONOO− cycle mechanism, if it applies to PAH, strong predictions that are not likely to be made based on any other unrelated hypothesis. Are they both correct? The following studies involve the ET-1 role in PAH but also in other tissues.
ET-1 has been shown to be induced by inflammatory cytokines [199–201]. It has also been shown to be induced by oxidants with such induction also lowered by antioxidants [202–204]. The activity of ET-1 has been shown to be increased by both NF-𝜅B action and also the action of another inflammatory cytokine activated pathway [205–207]. Thus, the inflammatory part of the NO/ONOO− cycle has a powerful role in increasing ET-1 activity. Furthermore BH4 was shown to substantially lower ET-1 levels [208], so that BH4 depletion, another part of the NO/ONOO− cycle has a role in raising ET-1 levels. ET-1 levels are also reported to be increased by TRPV1 [209, 210], another part of the cycle. It follows from these observations that elements of the NO/ONOO− cycle (at least five, in this case) have substantial roles in raising ET-1 levels, as predicted if PAH is a NO/ONOO− cycle disease and that ET-1 elevation also has a causal role in PAH.
Furthermore, ET-1 action has been shown to elevate, in turn, essentially the entire NO/ONOO− cycle. It increases oxidative stress [211–217], superoxide [211, 213–216], the ONOO− marker 3-nitrotyrosine [211, 212], NF-𝜅B activity [211, 217], and NO [211, 213,218], lowers levels of BH4 [213–215, 217, 219], stimulates iNOS induction [211, 217], increases TRP receptor function [220, 221], increases levels of intracellular calcium [222–224], increases NMDA activity [225, 226], produces mitochondrial dysfunction [227, 228], and increases inflammatory cytokine levels [206, 207, 217]. All of these provide evidence for elevation of each of the NO/ONOO− cycle elements.
However, there is one fact that requires some explanation. Even though there are studies showing that ET-1 increases iNOS induction [211, 217], there are also studies showing that it can decrease iNOS induction [219, 229]. These latter studies are in situations where substantial iNOS was already induced before ET-1 exposure and where lowered iNOS induction was accompanied by BH4 depletion [219, 229]. It has been shown that high level iNOS induction requires substantial pools of BH4 [230]. It is the author’s view, therefore, that this lowered iNOS induction is probably caused by the BH4 depletion produced by ET-1 and therefore that these observations do not conflict with the conclusion that ET-1 acts by elevating the NO/ONOO− cycle. ET-1 lowers induction of the enzyme GTP cyclohydrolase I, the rate limiting step in the production of BH4 [216], providing further evidence for a BH4 role here.
In conclusion, then, there is substantial evidence that elevated ET-1 can be induced by five elements of the NO/ONOO− cycle and that it can act, in turn, to elevate all of the NO/ONOO− cycle elements. It follows that ET-1 acts, at least in part, by elevating the NO/ONOO− cycle in PAH.
7. Elevated NO/ONOO− Cycle Elements in the Chronic Phase of PAH and Therapy via Agents that Lower NO/ONOO− Cycle Elements
The second principle of the NO/ONOO− cycle is that the elements of the cycle should be elevated in the chronic phase of the disease. And the fifth principle is that NO/ONOO−cycle diseases should respond to therapies that lower NO/ONOO− cycle elements and should, therefore, be treated in this way. The studies supporting both of these principles in PAH often deal with both of them, so both of these principles will be treated together here. We have already discussed substantial evidence for this in PAH in the context of the action of the various stressors that initiate PAH—evidence discussed in the previous two sections of this paper. Many of the studies cited in those two sections are not only relevant to the issue of disease initiation but also are relevant to the issue of the chronic phase of the disease. This section considers still additional studies that relate to these same issues, are elements of the cycle elevated in the chronic phase of PAH? Are agents that lower NO/ONOO− cycle elements helpful in therapy. Much of this discussion focuses on animal model studies.
Bowers et al. [28] was a histological study of human lung tissue in PAH, which showed that several markers of oxidative stress were elevated, that 3-nitrotyrosine, a marker of ONOO− was elevated and that inflammatory markers were elevated as well. They suggest that [28] chronic prostacyclin infusion, an anti-inflammatory treatment is helpful, suggesting that agents lowering the inflammatory part of the cycle may be useful in therapy.
Lakshminrusimha et al. [231] found that isoprostane, a marker of oxidative stress, and 3-nitrotyrosine, a marker of ONOO−, were both elevated in PAH and that addition of intratracheal superoxide dismutase (SOD) an enzyme that degrades superoxide, produced symptomatic improvement and lowered both of these markers. Interestingly, they also showed that inhaled NO produced symptomatic improvement but also raised ONOO−.
Herget et al. [232] reported increased NO production and elevated 3-nitrotyrosine in hypoxic PAH and also showed that antioxidants were helpful in prevention of PAH. This study, implicates NO, ONOO−, and oxidative stress and suggests that antioxidants may be helpful in therapy.
Weissmann et al. [233] reviewed evidence that both superoxide and its product, hydrogen peroxide, are elevated in PAH and argue that both mitochondrial and extramitochondrial mechanisms, including NADPH oxidase, are involved in superoxide generation. They also argue for a role of oxidant damage in causing the tissue remodeling in PAH.
Van Rheen et al. [185] studied normal mice and those expressing much higher levels of extracellular superoxide dismutase (SOD) in PAH initiated by bleomycin. The vascular remodeling and PAH were greatly lowered by SOD and survival was greatly improved. These studies provide strong support for the view that superoxide has an important role in tissue remodeling and PAH and that an agent that acts to lower superoxide is, therefore very helpful in prevention.
Dorfmüller et al. [234] showed that a marker of oxidative stress, malondialdehyde, 3-nitrotyrosine (ONOO− marker), and an inflammatory cytokine were all elevated in the tissue remodeling in PAH. There were also changes in expression of antioxidant enzymes, providing further support for a role of oxidative stress.
Rashid et al. [235] showed that a superoxide dismutase mimic greatly lowered the development of PAH, suggesting a causal role for superoxide and showing that an agent that lowers superoxide was very useful in prevention.
Lu et al. [236] showed that the drug, rosiglitazone, a PPAR𝛾 agonist, has favorable properties on human pulmonary artery smooth muscle, acting to lower two responses that are both elevated in response to hypoxia: increased NF-𝜅B activity and increased expression of one of the NADPH oxidase genes, Nox4. Rosiglitazone also acts to lower PAH responses. Because NADPH oxidase produced superoxide, this also implicates superoxide in PAH. These studies show that an agent that lowers both superoxide production and NF-𝜅B activity is helpful in therapy.
Oxidative stress and reactive oxygen species were discussed as causal factors in generating the symptoms and signs of PAH in the introduction to this paper [17, 22].
7.1. Peroxynitrite (ONOO−)
There are many papers, showing the 3-nitrotyrosine, a marker for ONOO− elevation, is raised in PAH, including four that were discussed above in this section [28, 185, 231,232]. In another study, Oishi et al. [237], showed that both superoxide and ONOO− were elevated following rebound from NO inhalation. So there should be no question that ONOO− is elevated in PAH. What is the main focus, here, therefore, is the study of acausal role of ONOO− in PAH.
Agbani et al. [238] showed that ONOO− stimulated the growth of pulmonary artery smooth muscle cells, a response involved in the remodeling response in PAH; the growth response was blocked by ebselen, an ONOO− scavenger, providing further evidence for a causal role for ONOO−.
Aggarwal et al. [239] showed that addition of an NO donor to pulmonary artery endothelial and smooth muscle cells increased ONOO− and increased nitration of protein kinase G. This response was shown to be blocked by either superoxide dismutase (SOD) or by an SOD mimic drug. This study followed an earlier one showing that nitration of the G kinase lowered its responsiveness to cGMP stimulation and therefore lowered its ability to produce NO-stimulated vasodilation and such attenuated vasodilation was also demonstrated in this study [239]. One part of the study showed that inhaled NO produced elevation of ONOO−, increased G kinase nitration, and decreased G kinase activity, providing a mechanism for lowered responsiveness to inhaled NO with time. Interestingly, in one part of the study [239], L-arginine was found to lower both ONOO− following simultaneous NO inhalation and also helped to maintain G kinase activity; this is important because L-arginine increases NO synthesis while lowering superoxide production from uncoupled nitric oxide synthases; the arginine finding argues, therefore, that superoxide generated on the uncoupled nitric oxide synthase enzyme may have a key role here. The causal role of ONOO− here is demonstrated by its known role in protein nitration, the demonstrated causation of lowered G kinase activity by a compound that breaks down to produce ONOO− and also the demonstrated causal role in this study of both of the ONOO− precursors, NO, and superoxide. This is a clear study demonstrating that an important change in the vasculature in PAH, namely, lowered responsiveness to NO mediated vasodilation, is caused by ONOO− mediated nitration of a particular protein, the G kinase. It also shows the efficacy of agents lowering superoxide.
In another study, Agbani et al. [240] demonstrated that ONOO− stimulates pulmonary endothelial cell and smooth muscle proliferation by a regulatory pathway involving both ERK and PKC protein kinases as well as two growth factor receptors. The causal role of ONOO− was demonstrated here, both by adding pure peroxynitrite (ONOO−) and by using the ONOO− scavenger ebselen to block these responses [240]. Belik et al. [241] asked whether hypoxia-caused PAH and vascular remodeling were caused by ONOO−elevation by using a ONOO− decomposition catalyst, FeTPPS. They showed that FeTPPS blocked some but not other changes [241], showing that those that were blocked were probably produced by ONOO−. Masood et al. [242] used this same FeTPPS decomposition catalyst to determine whether ONOO− was responsible for various changes in the pulmonary vasculature produced by 60% oxygen exposure in neonatal rats, leading to PAH. They concluded that some but not other changes were caused by ONOO−. It may be inferred from the above studies that many of the changes seen with PAH in the pulmonary vasculature are caused by ONOO−, the most central element in the NO/ONOO− cycle but that there are others that are not caused by ONOO− but perhaps may involve other cycle elements.
7.2. NF-𝜅B
One study discussed earlier in this section, Lu et al. [236] implicated elevated NF-𝜅B activity in PAH as did several studies cited in the previous two sections of the paper. There are several additional studies that implicate elevated NF-𝜅B activity in PAH. In one of these, Liu et al. [243] studied increased NF-𝜅B expression that developed along with PAH. The statin drug simvastatin was shown to have a favorable effect, apparently in part at least, by suppressing the increase in NF-𝜅B expression. A study by Li et al. [244] was not a study on PAH but was a study of the effect of an inflammatory protein found in PAH, C-reactive protein on the expression of NF-𝜅B and also the inflammatory cytokine IL-6 in pulmonary artery smooth muscle cells. They showed that both NF-𝜅B expression and IL-6 expression were raised by C-reactive protein and that these raises were suppressed by another statin drug, atorvastatin. Both of these studies provide some support for the view that increased NF-𝜅B expression and in the latter study, the cytokine IL-6, may have roles in PAH and that suppressing these roles with statin drugs may be useful therapeutically.
Another study suggesting a causal role of NF-𝜅B elevation is that of Kimura et al. [245], which showed that a nanoparticle-mediated delivery of an NF-𝜅B decoy (i.e., a nucleic acid-like sequence to which NF-𝜅B binds) to the lungs, greatly attenuated inflammation and smooth muscle proliferation in a monocrotaline PAH model. They also reported NF-𝜅B activity elevation in the lungs of patients with PAH.
Sawada et al. [246], studying a monocrotaline rat model of PAH, found prolonged elevation of NF-𝜅B activity. They used the NF-𝜅B inhibitor pyrrolidine dithiocarbamate and showed that it lowered NF-𝜅B activity and ameliorated the PAH development. Their results both confirm a role of NF-𝜅B elevation in PAH and suggest that agents lowering NF-𝜅B activity should be useful in therapy.
Huang et al. [247] did a similar study, also in a monocrotaline rat PAH model but got very different results. They found only a brief elevation of NF-𝜅B activity following monocrotaline treatment and did not confirm the lowering of NF-𝜅B activity by pyrrolidine dithiocarbamate. They state that the “reason for the discrepancy between our results and those of Sawada et al. is not clear.”
Two additional studies showed that NF-𝜅B activity can produce vascular smooth muscle proliferation [248, 249]. In one of these studies [248], antisense polynucleotides blocked expression of one of the protein subunits of NF-𝜅B and blocked vascular smooth muscle proliferation. In the other study [249], a hyperactive mutant gene for the inhibitor of NF-𝜅B IkappaBalpha, greatly lowered NF-𝜅B activity and also blocked vascular smooth muscle proliferation. These studies show that vascular smooth muscle proliferation can be caused by NF-𝜅B activity elevation and also lowered by agents that lower NF-𝜅B activity.
In summary, there is substantial evidence that elevated NF-𝜅B activity has a role in PAH and that it has a causal role in the development of PAH, being specifically implicated in smooth muscle proliferation and therefore in tissue remodeling.
7.3. Inflammatory Cytokines
There have been numerous studies reporting elevated levels of inflammatory cytokines in PAH, including two discussed above in this section [234, 243]. A number of such studies reported elevation of multiple inflammatory cytokines/chemokines in PAH. For example, Soon et al. [250] reported that IL-1𝛽, −2, −4, −6, −8, −10, and −12p70, as well as TNF-𝛼were elevated in PAH patients. Elevated levels of four of these were associated with lowered survival, with IL-6 showing the highest such association. Li et al. [251] found that 11 inflammatory proteins were elevated in adventitial fibroblasts from calves suffering from hypoxic PAH, most of these proteins different from the ones studied by Soon et al. [250], but including IL-1𝛽 and IL-6. Yu et al. [252] reported elevated levels of IL-1𝛽, TNF-𝛼, and IL-6 in PAH patients undergoing hemodialysis. Sin and Man [253] also provided evidence for elevation of IL-1𝛽, TNF-𝛼, and IL-6 in PAH patients. Hamal et al. [254] showed that PAH induced by injection of microparticulate cellulose into susceptible chickens was accompanied by elevated expression of several inflammatory cytokines including IL-1𝛽, IL-4, IL-6, IL-8, and IFN𝛾. An IL-1 receptor antagonist was shown to lower PAH responses, suggesting a causal role for IL-1𝛽 elevation in PAH [255]. Wanderer provided a strong rationale for using IL-1 𝛽 antagonists or receptor blockers in PAH treatment [256].
A number of studies have focused, to some extent, specifically on a role for IL-6 in PAH. Kalambokis et al. [257] reported high serum levels of IL-6 in cirrhosis-associated PAH. Chaouat et al. [258] found that COPD patients with PAH had higher levels of IL-6 than did COPD patients without PAH. They also showed that individuals carrying a genetic polymorphism in the IL-6 gene associated with higher gene expression had significantly higher prevalence of PAH, strongly suggesting a causal role for IL-6. Finally they showed [258] that pulmonary artery smooth muscle cells exposed to hypoxic conditions had twice the IL-6 mRNA as those not exposed to hypoxic conditions. The concluded that “inflammation, most likely involving IL-6, may contribute substantially to PH complicating COPD.” Furuya et al. [259] reviewed evidence implicating IL-6 in promoting smooth muscle and epithelial proliferation in PAH. They showed that a patient with severe refractory PAH responded dramatically to treatment with a humanized monoclonal antibody to the IL-6 receptor and suggest that IL-6 blockade may be a promising adjunct treatment for PAH. Steiner et al. [260] compared transgenic mice overexpressing IL-6 with normal mice. They found that such transgenic mice differed from normals under both hypoxic and normoxic conditions, leading to increased occurrence of a number of changes characteristic of PAH. They suggest that “IL-6 promotes the development and progression of pulmonary vascular remodeling and PAH through proproliferative and antiapoptotic mechanisms.”
It may be seen from the above two paragraphs, that there are wide ranging proinflammatory changes associated with PAH and that IL-6 elevation may play a particularly important role in PAH development. One study [259] suggests that IL-6 action may be an important therapeutic target in PAH and two other studies [255, 256] suggest that IL-1𝛽 action may be as well.
7.4. iNOS Induction
iNOS induction has been reported in two additional studies to be implicated in PAH [261, 262]. In one of these [262], a specific inhibitor of iNOS was shown to greatly lower PAH development, arguing for a causal role of iNOS induction in PAH. An iNOS knockout mouse was less sensitive to exacerbation of PAH, including inflammatory responses, as discussed above [263].
7.5. Mitochondrial Dysfunction
Quite a number of studies have reported substantial mitochondrial dysfunction in PAH [264–269]. The genetic studies in [266–268] strongly suggest that such mitochondrial dysfunction can have a strong, causal role in PAH and one study used mitochondrial inhibitors which showed a complex causal role in PAH [269]. Four of these studies [264,266, 268, 269] all suggest mechanisms by which mitochondrial dysfunction may contribute to the symptoms and signs of PAH. However, none of these provide any evidence that agents that improve mitochondrial function can prevent or produce improvements in PAH. This last issue of possible therapy via improved mitochondrial seems to be a neglected one in PAH. The only such studies that the author is familiar with are two studies showing that inhibitors of poly (ADP-ribose) polymerase (or synthetase) (PARS or PARP) provide substantial protection from PAH [270, 271]. This PARP activity is known to initiate one of the two cascades of events, leading to mitochondrial dysfunction, proposed to be part of the NO/ONOO− cycle, and therefore lowering its activity will be predicted to have a major effect in lowering mitochondrial dysfunction, as has been shown in a number of chronic inflammatory diseases.
7.6. TRP Receptors and Also Intracellular Calcium
The study of Yu et al. [272] showed that a single nucleotide polymorphism in the TRPC6 gene, introducing an NF-𝜅B site in the enhancer region of the gene, increased susceptibility to PAH. This study implicates both a TRP receptor and consequent increased intracellular calcium in PAH. Two studies, in addition to the ones cited above, also suggest roles of the TRP group of receptors and increased intracellular calcium in PAH [273, 274]. Capsaicin is a TRPV1 agonist which over the longer term, produces very substantial down-regulation of the TRPV1 and several other TRP receptors. It is interesting, therefore that several studies showed that capsaicin pretreatment over a period of days produces greatly decreased PAH induction [275–277].
There are a variety of additional studies showing that elevated intracellular calcium levels have causal roles in PAH [273–282], including studies using calcium channel blockers [279–282] to obtain clinical improvement. It follows that agents lowering intracellular calcium may also be useful in therapy. A role for intracellular calcium is further supported by a study showing that inhibition of calcium-dependent protease, calpain blocked the development of several features of PAH [283].
7.7. NO Elevation?
The NO/ONOO− cycle mechanism predicts that NO synthesis will be increased through iNOS induction and by calcium-stimulation of eNOS and nNOS activity but that it will also be decreased through BH4 depletion and consequent partial uncoupling of all three NOSs [8]. Furthermore, as discussed above, BH4 depletion can lead to lowered eNOS and iNOS expression. Consequently, it is unclear whether the cycle mechanism should predict whether an increase or decrease in NO should be expected. However, most proposed NO/ONOO− cycle diseases have published studies showing apparent increases in NO synthesis.
It is important to raise the question as to whether NO should be considered an element of the cycle or not. Clearly, the answer is yes, if one considers that NO is essential for the production of peroxynitrite (ONOO−), but the answer is less clear as to whether its synthesis must be elevated in NO/ONOO− cycle diseases. These considerations should be kept in mind in considering the data on NO in PAH.
Most studies of NO in PAH patients have measured exhaled NO levels from the lungs in order to assess possible changes in NO synthesis in PAH [284–290]. NO is relatively stable in the gas phase, allowing for such measurements, whereas the instability of NO in biological fluids means that estimates of NO synthesis from blood measurements are usually done via nitrate/nitrite products derived from NO. However the instability of NO in biological fluids means that anything that lowers the movement of NO from the pulmonary arteries into the gas phase, such as fibrosis, arterial remodeling, or lowered ciliary activity may be expected to lower measured exhaled NO. Furthermore, the reaction of NO with superoxide to form ONOO− (both superoxide and ONOO− are clearly both elevated in PAH), will also lower any measurement of exhaled NO.
The majority of studies on exhaled NO in PAH patients report lowered levels when compared with controls [284–287, 289], but two such studies report apparent elevated levels, with one reporting a nonsignificant trend [288] and the other significant elevation [290]. Consequently, there appears to be substantial variation in these studies. Two of these studies compared exhaled NO levels with nitrate/nitrite levels in the blood in the same individuals. Both found that although exhaled NO levels were lower in PAH patients, blood levels of nitrite/nitrate were elevated [284, 289]. The interpretations of these results by the authors of these two studies were different from one another, but clearly one interpretation is the one suggested in the previous paragraph, that NO synthesis may be elevated but that movement of NO from the pulmonary arteries to the gas phase may be substantially slowed in PAH. If this interpretation is correct, it raises questions about whether any of the studies of lowered exhaled NO in PAH should be interpreted as good measurements of NO synthesis. In addition, three studies reported that therapies that produced symptomatic improvement in PAH patients, lowered exhaled NO levels [285, 291, 292], suggesting a correlative relationship between lowered NO and improved symptoms.
The observations discussed in the previous two paragraphs show that NO is not likely to be consistently low in PAH and that correlative data suggest that therapies lowering NO can produce therapeutic improvement, although none of these therapies were designed to lower NO. Before leaving this issue, it is important to discuss an additional type of observation on PAH. It has often been reported that lowered eNOS expression is found in the pulmonary arteries and associated cells in PAH, and such observations have often been used to suggest that low NO helps cause PAH. However, it should be noted that BH4 depletion, a part of the cycle that has been confirmed in PAH, causes lowered expression of eNOS (reviewed in [193]). It is possible, therefore, that such lowered eNOS expression is a protective mechanism, aimed at avoiding excessive superoxide production, rather than a causal mechanism in PAH; if this view is correct, then it follows that this protective mechanism is insufficient to avoid PAH under conditions where the disease is induced.
In conclusion, then, none of the studies on NO in PAH can be argued to conflict with the NO/ONOO− cycle mechanism, although like many other types of data, there are multiple interpretations of these data that can be suggested.
One of the questions that must be raised in the context of a possible NO/ONOO− cycle mechanism for PAH is what role NO has in the process? Clearly inhaled NO can produce an initial lowering of hypertension, something that might be expected, based on the role of NO as a vasodilator and also possibly the action of NO in lowering two NO/ONOO−cycle elements, NF-𝜅B activity and NMDA activity. However, the most crucial NO role in the cycle is acting as a precursor for ONOO− and consequently a pathophysiological role for NO is an essential part of the cycle. Such a pathophysiological role for NO in PAH is most clearly seen in the rebound that occurs after NO inhalation, when NO is no longer producing favorable responses and therefore is no longer inhaled. The rebound response to previous NO inhalation clearly shows that NO can produce a chronic exacerbation of the disease and clearly establishes such a pathophysiological role. However, a question should be raised is whether the properties of that chronic exacerbation are consistent with that predicted by the NO/ONOO− cycle There are a number of studies that suggest that they are. For example, Weinberger et al. [293] showed that previous NO exposure led not only to elevated ONOO−, as predicted by the NO precursor role in ONOO−formation, but also increased superoxide production and iNOS activity, consequences that must be indirect but are predicted by the cycle. Wedgwood et al. [212] showed a role for both increased ONOO− and superoxide in the rebound response following NO inhalation. Shanley et al. [263] showed inflammatory responses following NO inhalation, responses that were greatly lowered in an iNOS knockout mouse, showing that such inflammatory responses were not only produced by such NO inhalation, but were lowered by blocking an important part of the NO/ONOO− cycle, namely iNOS induction. The Lakshminrusimha et al. study [231] showed that inhaled NO produced increases in ONOO−. The Aggarwal et al. study [239] showed that inhaled NO not only raised ONOO−, but also was ONOO− dependent nitration of the G kinase enzyme, leading to lowered vasodilation, an important part of the rebound phenomenon. Oishi et al. [237] found that both ONOO− and superoxide were elevated following NO inhalation and established a causal role of superoxide in the rebound response by using polyethylene glycol-conjugated SOD to inhibit the rebound response.
All of these findings are consistent with upregulation of the NO/ONOO− cycle as a consequence of inhaled NO, with NO acting as a ONOO− precursor. The knockout iNOS mouse study suggests that lowering NO can be helpful and can lower the inflammatory parts of the cycle.
This does not mean that there are no situations where NO can be helpful, certainly its role in vasodilation and its potential role in lowering both NMDA receptor activity and NF-𝜅B activity can also be helpful, but in a NO/ONOO− cycle situation, its role as a precursor of ONOO− must be considered to be paramount.
7.8. BH4 Therapy
Several studies implicating BH4 depletion in PAH were discussed in the previous two sections of this paper. Three studies suggest that agents that raise BH4 levels may be helpful in therapy. One was a study on the safety of sapropterin (the drug name for BH4) in PAH patients [294].
In another paper, Teng et al. [295] studied the pulmonary artery endothelial tissues from fetal lambs with pulmonary hypertension comparing these is normal lambs. In this study Teng et al. [295] showed that the BH4 levels in the PAH lambs were very low compared with normals, leading to eNOS uncoupling, lowered NO and raised superoxide. Interestingly, the lowered BH4 levels were not only produced by ONOO− oxidation of BH4, but also by lowered GTP cyclohydrolase I, the rate limiting enzyme in BH4 synthesis. Addition of sepiapterin, a BH4 precursor to these PAH arterial tissues, raised BH4, raised NO synthesis and lowered superoxide. It also led to increased probable stability of the eNOS enzyme by increased association with Hsp90 protein. Most importantly it help restore angiogenesis which is deficient in PAH. The authors suggest that raising BH4 levels is important in PAH therapy.
A third study showed that heat treatment, such as that found in sauna therapy, was therapeutically useful in PAH patients [296]. Sauna therapy is thought to act by raising the level of the rate limiting enzyme in BH4 synthesis, GTP cyclohydrolase I, and therefore raising BH4 availability [297].
While none of these studies can be taken as definitive, they suggest that raising BH4 levels is likely to be useful in PAH therapy.
7.9. NMDA Activity?
Evidence suggesting excessive activity of the NMDA receptors in PHA, discussed above such a mechanism in case initiation by endotoxin exposure, elevated homocysteine or paraquat exposure, exacerbation by ammonia, or the evidence, also discussed above, showing that ET-1 raised NMDA activity. Each of these observations are susceptible to various interpretations, however. Similarly, while there have been no studies aimed at testing the efficacy of agents lowering NMDA activity in PAH, five studies have shown the value of raising magnesium levels in PAH [298–303]. The NMDA receptors are highly susceptible to being activated in conditions of even marginal magnesium deficiency, so an attractive interpretation of the magnesium studies is that magnesium is acting by lowering NMDA activity. Other interpretations are possible here, as well. Clearly, this is the part of the cycle that is most weakly linked to PAH and it may be argued that it may not have a causal role at all.
8. Is Resveratrol a Magic Bullet for the Treatment of PAH?
Some very exciting research has shown that an agent that gives very good clinical responses in preventing PAH acts to down-regulate most of the NO/ONOO− cycle elements. This research was performed by Csiszar, Ungvari, and their colleagues at New York Medical College. In one study [304], they showed the following NO/ONOO− cycle elements were elevated in PAH: oxidative stress, the inflammatory cytokines IL-6 and TNF-𝛼, other inflammatory genes, NF-𝜅B and iNOS induction. All of these were blocked along with the PAH by resveratrol, the stilbene flavonoid reported to be a wide ranging health-promoting agent. In another study [305] resveratrol prevented PAH development along with elevation of three cytokines, oxidative stress, endothelial dysfunction, and upregulation of NADPH oxidase, one of the mechanisms that generates superoxide. In a third study [306], this one on coronary arterial endothelial cells, resveratrol improved mitochondrial function, increased the mitochondrial SOD (an enzyme that gets rid of superoxide), increased reduced glutathione (an important antioxidant mechanism), lowered oxidative stress, lowered mitochondrial production of reactive oxygen species, all favorable changes lowering NO/ONOO− cycle elements. A study by another research group [307] showed that resveratrol increased mitochondrial biogenesis and lowered angiotensin-II-dependent activity, thus providing improvements via two distinct mechanisms. Another group [308] showed that resveratrol increases synthesis of all three superoxide dismutases (not just the mitochondrial enzyme), lowered NADPH oxidase (an important source of superoxide), lowered superoxide (not surprisingly), lowered oxidative stress, and lowered ONOO−. Most importantly, they also showed that the synthesis of BH4 was increased by inducing increases levels of the enzyme GTP cyclohydrolase I, the rate limiting enzyme in the de novo pathway for BH4 synthesis. Not surprisingly, the increased BH4 led to increased coupling of the eNOS enzyme [308]. Other studies have shown that resveratrol decreases the activity of the NMDA receptors and also the related kainate receptors, acting on both the receptor activities themselves [309–311] and also acting to increase glutamate transport [312], thus lowering the extracellular glutamate that acts as an NMDA agonist. In each of these actions, resveratrol is thought to act by stimulating the activity of SIRT1, a protein that has a wide ranging activity in regulating gene expression. All of this suggests that resveratrol is almost a dream agent for lowering the NO/ONOO− cycle. By raising SIRT1, it lowers, at least in some tissues each of the following NO/ONOO− cycle elements: oxidative stress, ONOO−, mitochondrial dysfunction, superoxide, inflammatory cytokines and other inflammatory markers, NF-𝜅B and excessive NMDA activity while raising BH4 levels. Almost the whole cycle is lowered by resveratrol.
Should we be surprised that another research group asked [313]: is resveratrol the magic bullet for pulmonary hypertension?
It is this author’s view that resveratrol is likely to be a useful therapeutic agent for PAH but that it is not likely to be a magic bullet. The basis of this assessment is two-fold. Firstly, it is much more difficult, in general, to cure disease than to prevent it and this is to be especially expected for such a mechanism as the NO/ONOO− cycle, whose robust structure (Figures 1(a)–1(e)) strongly argues that it is difficult to down-regulate. These studies were basically preventative rather than curative [304–306]. Furthermore, the activity of SIRT1 [314], which mediates each of these resveratrol effects, is sensitive to oxidative inactivation and is also dependent on NAD but NAD can be massively depleted by the elevation of PARP activity by ONOO– in the NO/ONOO– cycle (see number 18 of the 34 mechanisms). It follows from this that repleting NAD with high doses of nicotinamide and nicotinic acid (forms of niacin) will be needed as well as the use of antioxidants and lowering of ONOO−, for resveratrol to be very active. In general, the more severe the disease and the cycle, the more difficult it will be to do all that effectively.
9. Discussion and Conclusions
The NO/ONOO− cycle is a complex biochemical/physiological vicious cycle that can explain various chronic inflammatory diseases localized to certain regions of the body. It should be considered as an etiologic mechanism only for such inflammatory diseases that can be initiated by stressors, including short-term stressors, that can elevate cycle elements and can, therefore, at least in principle, initiate the cycle through such elevation. Clearly PAH is such a disease and, therefore, PAH may be a candidate for such an etiology. What should be clear from this paper, is that there are many diverse studies on PAH, each implicating NO/ONOO− cycle elements in PAH (much of these are summarized in Table 2). While many of these do not show that these elements have causal roles in causing PAH, so that they may simply be an epiphenenon simply based on those studies, there are also many studies that do show a causal role for 11 out of 12 cycle elements. These collectively therefore provide strong support for a NO/ONOO− cycle etiology.
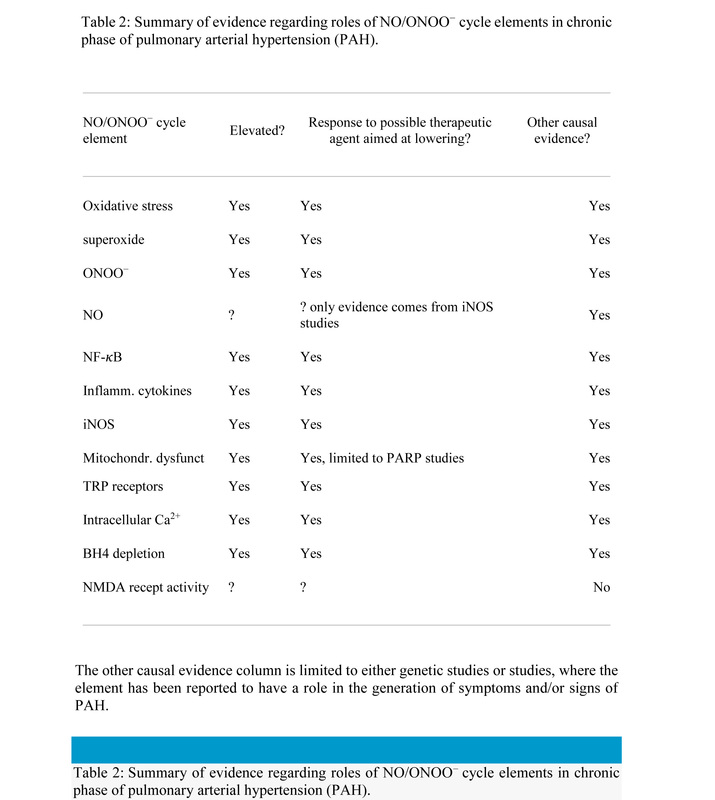
Furthermore, the cycle is based on five principles the fit to each of which provides a very different type of evidence for the causality of the cycle. In PAH, there is strong evidence that stressors that initiate cases of PAH elevate cycle elements, showing the each of these initiators can potentially act to initiate the cycle in this way (Principle 1), see Table 1. There is strong evidence that most of the elements of the cycle are elevated in the chronic phase of the disease (Principle 2). There is strong evidence that the symptoms and signs of PAH can be produced by cycle elements (Principle 3), provided in the introduction and elsewhere in this paper. There is also strong evidence for a local mechanism in PAH, localized to the pulmonary arteries (Principle 4), also discussed in the introduction. In addition, there is also strong evidence, mostly derived from animal model studies, that agents that lower cycle elements can be useful in therapy (Principle 5).
However, this does not mean that there are no weaknesses in the case. The weakest part of the argument is that there is no direct evidence that excessive NMDA activity in the pulmonary arteries have a role. There is evidence for the existence of NMDA receptors in the pulmonary arteries. There is evidence that two stressors that elevate NMDA activity, high levels of both homocysteine and ammonia, each have roles in causing PAH. However, there have been no studies to test whether the NMDA receptors have roles in these or other observations about PAH.
With regard to causal roles in PAH, the following elements of the cycle appear to have causal roles, as documented in various parts of this paper.(1)From the introduction. elevated ONOO−, consequent oxidative stress, inflammatory responses, and mitochondrial dysfunction all have causal roles in generating the symptoms and signs of PAH.(2)From the subsection on RhoA elevation, NF-𝜅B elevation, inflammatory cytokines, and oxidative stress all have causal roles in elevating RhoA.(3)From other sections on initiation of cases of PAH by initiating stressors, BH4 depletion, oxidative stress, two of the TRP receptors, and one of the inflammatory cytokines, all have causal roles. Independent of the issue of causality, each of the initiators, as seen in Table 1, elevate, multiple cycle elements.(4)From the section on Endothelin-1 (ET-1), the following have causal roles in raising ET-1 levels: NF-𝜅B, oxidative stress, inflammatory cytokines, and one of the TRP receptors (TRPV1).(5)Finally, from the section on elevated cycle elements in the chronic phase of disease and also therapy via agents that lower cycle elements 10 of the 12 cycle elements have causal roles based on therapy and 11 of the 12 have causal roles based on other types of evidence. The only element not causally implicated is excessive NMDA activity where no directly relevant studies are available. There are, in addition many other studies where these cycle elements are implicated but where causality is uncertain. It is difficult to see how that could all be true unless the NO/ONOO− cycle or something similar to it is the central cause of PAH.
There is one additional consideration with regard to these data. Each of the individual observations summarized earlier in this Discussion and Conclusions Section can be interpreted in various ways. It is the pattern of evidence that argues for a NO/ONOO−cycle etiology, not the individual studies or observations. And in most cases, this is true of other proposed NO/ONOO− cycle diseases as well—it is only the pattern of evidence that makes a convincing case. However in PAH there are two exceptions to this and they relate to biological components that have causal roles in PAH.
As discussed in the Endothelin-1 (ET-1) section, there is strong evidence that ET-1 has a causal role in PAH. The only way that ET-1 can have a causal role if PAH is a NO/ONOO− cycle disease, is if ET-1 is behaving as part of the cycle in the pulmonary arteries. That is, ET-1 must be synthesized in increasing amounts in response to cycle elements and must in turn increase cycle elements. These are, then, strong predictions of the NO/ONOO− cycle mechanism, if it applies to PAH, predictions that are not likely to be made based on any other hypothesis. So are both of the predictions true? Evidence was already summarized in 4 above that ET-1 is synthesized in response to four cycle elements, NF-𝜅B oxidative stress, inflammatory cytokines, and one of the TRP receptors (TRPV1). So the first prediction is clearly supported. Going back to the Endothelin-1 section, it can be seen that ET-1 acts, in turn, to elevate the following elements of the cycle: oxidative stress, superoxide, the ONOO− cycle marker 3-nitrotyrosine, NF-𝜅B activity, BH4 depletion, iNOS induction, TRP receptor function, intracellular calcium levels, mitochondrial dysfunction, NMDA activity, and inflammatory cytokines. Essentially the whole cycle is elevated in response to ET-1. I must note, that some of these studies have been done not in the context of PAH but rather in other regions of the body where ET-1 is also active, including regions of the brain. Nevertheless, they provide about the closest thing to smoking gun evidence that one is likely to find, confirming specific predictions of a NO/ONOO− cycle etiology for PAH.
There is a similar, albeit somewhat weaker, case for a causal role for RhoA as a causal factor in PAH acting as a NO/ONOO− cycle element. Three cycle elements act to raise RhoA, as seen in 2 above. RhoA acts in turn, as shown in that section of the paper, to elevate superoxide, ONOO−, oxidative stress, BH4 depletion and intracellular calcium. So here we have a similar, albeit slightly weaker example of smoking gun evidence for a NO/ONOO− cycle etiology for PAH.
It may be concluded that it is highly probable that PAH is a NO/ONOO− cycle disease, with the only substantial weakness being the lack of direct evidence for a causal role for excessive NMDA activity in PAH.
This substantial weakness provides a simple test of the NO/ONOO− cycle mechanism for PAH. The cycle mechanism predicts that excessive NMDA activity has an important causal role in PAH and that this role may be particularly important in initiation via mechanisms such as hypoxia, homocysteine, or endotoxin, which, as discussed above, may act in part via excessive NMDA activity. These are easily testable predictions and should be tested in my no doubt somewhat biased view, possibly by using such relatively well-tolerated NMDA antagonists such as memantine or dextromethorphan.
An inference from the NO/ONOO− cycle mechanism for PAH is that it is very important to research multiagent protocols for PAH treatment, with agents acting to down-regulate different parts of the cycle. The robust nature of the cycle, as seen in Figures 1(a)–1(e)strongly suggest that such multiagent protocols may be the only approach to get a good sustained clinical response and possibly a cure.
The NO/ONOO− cycle concept comes from a group of previously unexplained diseases including chronic fatigue syndrome, multiple-chemical sensitivity, and fibromyalgia and also from the 34 well-accepted mechanisms listed and documented in the third section of this paper. In science, it is always important to distinguish data that were used to formulate a theory from other data that can be used to independently test it. Clearly one way of doing this for the NO/ONOO− cycle as a possibly widely applicable disease paradigm is to determine how well other diseases fit the predictions of this theory. Because of the excellent fit of PAH, it joins tinnitus [11] as an important independent test of the NO/ONOO− cycle theory of disease.
References
However, this does not mean that there are no weaknesses in the case. The weakest part of the argument is that there is no direct evidence that excessive NMDA activity in the pulmonary arteries have a role. There is evidence for the existence of NMDA receptors in the pulmonary arteries. There is evidence that two stressors that elevate NMDA activity, high levels of both homocysteine and ammonia, each have roles in causing PAH. However, there have been no studies to test whether the NMDA receptors have roles in these or other observations about PAH.
With regard to causal roles in PAH, the following elements of the cycle appear to have causal roles, as documented in various parts of this paper.(1)From the introduction. elevated ONOO−, consequent oxidative stress, inflammatory responses, and mitochondrial dysfunction all have causal roles in generating the symptoms and signs of PAH.(2)From the subsection on RhoA elevation, NF-𝜅B elevation, inflammatory cytokines, and oxidative stress all have causal roles in elevating RhoA.(3)From other sections on initiation of cases of PAH by initiating stressors, BH4 depletion, oxidative stress, two of the TRP receptors, and one of the inflammatory cytokines, all have causal roles. Independent of the issue of causality, each of the initiators, as seen in Table 1, elevate, multiple cycle elements.(4)From the section on Endothelin-1 (ET-1), the following have causal roles in raising ET-1 levels: NF-𝜅B, oxidative stress, inflammatory cytokines, and one of the TRP receptors (TRPV1).(5)Finally, from the section on elevated cycle elements in the chronic phase of disease and also therapy via agents that lower cycle elements 10 of the 12 cycle elements have causal roles based on therapy and 11 of the 12 have causal roles based on other types of evidence. The only element not causally implicated is excessive NMDA activity where no directly relevant studies are available. There are, in addition many other studies where these cycle elements are implicated but where causality is uncertain. It is difficult to see how that could all be true unless the NO/ONOO− cycle or something similar to it is the central cause of PAH.
There is one additional consideration with regard to these data. Each of the individual observations summarized earlier in this Discussion and Conclusions Section can be interpreted in various ways. It is the pattern of evidence that argues for a NO/ONOO−cycle etiology, not the individual studies or observations. And in most cases, this is true of other proposed NO/ONOO− cycle diseases as well—it is only the pattern of evidence that makes a convincing case. However in PAH there are two exceptions to this and they relate to biological components that have causal roles in PAH.
As discussed in the Endothelin-1 (ET-1) section, there is strong evidence that ET-1 has a causal role in PAH. The only way that ET-1 can have a causal role if PAH is a NO/ONOO− cycle disease, is if ET-1 is behaving as part of the cycle in the pulmonary arteries. That is, ET-1 must be synthesized in increasing amounts in response to cycle elements and must in turn increase cycle elements. These are, then, strong predictions of the NO/ONOO− cycle mechanism, if it applies to PAH, predictions that are not likely to be made based on any other hypothesis. So are both of the predictions true? Evidence was already summarized in 4 above that ET-1 is synthesized in response to four cycle elements, NF-𝜅B oxidative stress, inflammatory cytokines, and one of the TRP receptors (TRPV1). So the first prediction is clearly supported. Going back to the Endothelin-1 section, it can be seen that ET-1 acts, in turn, to elevate the following elements of the cycle: oxidative stress, superoxide, the ONOO− cycle marker 3-nitrotyrosine, NF-𝜅B activity, BH4 depletion, iNOS induction, TRP receptor function, intracellular calcium levels, mitochondrial dysfunction, NMDA activity, and inflammatory cytokines. Essentially the whole cycle is elevated in response to ET-1. I must note, that some of these studies have been done not in the context of PAH but rather in other regions of the body where ET-1 is also active, including regions of the brain. Nevertheless, they provide about the closest thing to smoking gun evidence that one is likely to find, confirming specific predictions of a NO/ONOO− cycle etiology for PAH.
There is a similar, albeit somewhat weaker, case for a causal role for RhoA as a causal factor in PAH acting as a NO/ONOO− cycle element. Three cycle elements act to raise RhoA, as seen in 2 above. RhoA acts in turn, as shown in that section of the paper, to elevate superoxide, ONOO−, oxidative stress, BH4 depletion and intracellular calcium. So here we have a similar, albeit slightly weaker example of smoking gun evidence for a NO/ONOO− cycle etiology for PAH.
It may be concluded that it is highly probable that PAH is a NO/ONOO− cycle disease, with the only substantial weakness being the lack of direct evidence for a causal role for excessive NMDA activity in PAH.
This substantial weakness provides a simple test of the NO/ONOO− cycle mechanism for PAH. The cycle mechanism predicts that excessive NMDA activity has an important causal role in PAH and that this role may be particularly important in initiation via mechanisms such as hypoxia, homocysteine, or endotoxin, which, as discussed above, may act in part via excessive NMDA activity. These are easily testable predictions and should be tested in my no doubt somewhat biased view, possibly by using such relatively well-tolerated NMDA antagonists such as memantine or dextromethorphan.
An inference from the NO/ONOO− cycle mechanism for PAH is that it is very important to research multiagent protocols for PAH treatment, with agents acting to down-regulate different parts of the cycle. The robust nature of the cycle, as seen in Figures 1(a)–1(e)strongly suggest that such multiagent protocols may be the only approach to get a good sustained clinical response and possibly a cure.
The NO/ONOO− cycle concept comes from a group of previously unexplained diseases including chronic fatigue syndrome, multiple-chemical sensitivity, and fibromyalgia and also from the 34 well-accepted mechanisms listed and documented in the third section of this paper. In science, it is always important to distinguish data that were used to formulate a theory from other data that can be used to independently test it. Clearly one way of doing this for the NO/ONOO− cycle as a possibly widely applicable disease paradigm is to determine how well other diseases fit the predictions of this theory. Because of the excellent fit of PAH, it joins tinnitus [11] as an important independent test of the NO/ONOO− cycle theory of disease.
References
- M. L. Pall, Explaining,“Unexplained Illnesses”: Disease Paradigm for Chronic Fatigue Syndrome, Multiple Chemical Sensitivity, Fibromyalgia, Post-Traumatic Stress Disorder, Gulf War Syndrome and Others, Harrington Park Press, New York, NY, USA, 2007.
- M. L. Pall, “Multiple chemical sensitivity: toxicological questions and mechanisms,” in General and Applied Toxicology, B. Ballantyne, T. C. Marrs, and T. Syversen, Eds., pp. 2303–2352, John Wiley and Sons, London, UK, 3rd edition, 2009. View at Google Scholar
- M. L. Pall, “The NO/ONOO− cycle mechanism as the cause of chronic fatigue syndrome/myalgia encephalomyelitis,” in Chronic Fatigue Syndrome: Symptoms, Causes and Prevention, E. Svoboda and K. Zelenjcik, Eds., chapter 2, Nova, New York, NY, USA, 2009. View at Google Scholar
- M. L. Pall, “The NO/ONOO− cycle as the cause of fibromyalgia and related illnesses: etiology, explanation and effective therapy,” in New Research in Fibromyalgia, J. A. Pederson, Ed., chapter 2, pp. 39–59, Nova Science, New York, NY, USA, 2006. View at Google Scholar
- M. L. Pall, “Elevated, sustained peroxynitrite levels as the cause of chronic fatigue syndrome,” Medical Hypotheses, vol. 54, no. 1, pp. 115–125, 2000. View at Publisher · View at Google Scholar · View at Scopus
- M. L. Pall, “Common etiology of posttraumatic stress disorder, fibromyalgia, chronic fatigue syndrome and multiple chemical sensitivity via elevated nitric oxide/peroxynitrite,” Medical Hypotheses, vol. 57, no. 2, pp. 139–145, 2001. View at Publisher · View at Google Scholar · View at Scopus
- M. L. Pall and J. D. Satterlee, “Elevated nitric oxide/peroxynitrite mechanism for the common etiology of multiple chemical sensitivity, chronic fatigue syndrome, and posttraumatic stress disorder,” Annals of the New York Academy of Sciences, vol. 933, pp. 323–329, 2001. View at Google Scholar · View at Scopus
- M. L. Pall, “Nitric oxide synthase partial uncoupling as a key switching mechanism for the NO/ONOO− cycle,” Medical Hypotheses, vol. 69, no. 4, pp. 821–825, 2007.View at Publisher · View at Google Scholar · View at Scopus
- M. L. Pall, “NMDA sensitization and stimulation by peroxynitrite, nitric oxide, and organic solvents as the mechanism of chemical sensitivity in multiple chemical sensitivity,” The FASEB Journal, vol. 16, no. 11, pp. 1407–1417, 2002. View at Publisher · View at Google Scholar · View at Scopus
- M. L. Pall, “How can we cure NO/ONOO− cycle diseases? Approaches to curing chronic fatigue syndrome/myalgic encephalomyelitis, fibromyalgia, multiple chemical sensitivity, Gulf War syndrome and possibly many others,” Townsend Letter for Doctors and Patients, pp. 75–84, 2010. View at Google Scholar
- M. L. Pall and S. A. Bedient, “The NO/ONOO− cycle as the etiological mechanism of tinnitus,” International Tinnitus Journal, vol. 13, no. 2, pp. 99–104, 2007. View at Google Scholar · View at Scopus
- J. C. Romero and J. F. Reckelhoff, “Role of angiotensin and oxidative stress in essential hypertension,” Hypertension, vol. 34, no. 4, pp. 943–949, 1999. View at Google Scholar · View at Scopus
- R. A. Cohen and X. Tong, “Vascular oxidative stress: the common link in hypertensive and diabetic vascular disease,” Journal of Cardiovascular Pharmacology, vol. 55, no. 4, pp. 308–316, 2010. View at Publisher · View at Google Scholar · View at Scopus
- T. J. Guzik, S. Mussa, D. Gastaldi et al., “Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase,” Circulation, vol. 105, no. 14, pp. 1656–1662, 2002. View at Publisher · View at Google Scholar · View at Scopus
- T. Münzel, T. Gori, R. M. Bruno, and S. Taddei, “Is oxidative stress a therapeutic target in cardiovascular disease?” European Heart Journal, vol. 31, no. 22, pp. 2741–2748, 2010. View at Publisher · View at Google Scholar · View at Scopus
- S. Milstien and Z. Katusic, “Oxidation of tetrahydrobiopterin by peroxynitrite: implications for vascular endothelial function,” Biochemical and Biophysical Research Communications, vol. 263, no. 3, pp. 681–684, 1999. View at Publisher ·View at Google Scholar · View at Scopus
- P. Crosswhite and Z. Sun, “Nitric oxide, oxidative stress and inflammation in pulmonary arterial hypertension,” Journal of Hypertension, vol. 28, no. 2, pp. 201–212, 2010. View at Publisher · View at Google Scholar · View at Scopus
- L. J. Janssen, “Isoprostanes and lung vascular pathology,” American Journal of Respiratory Cell and Molecular Biology, vol. 39, no. 4, pp. 383–389, 2008. View at Publisher · View at Google Scholar · View at Scopus
- R. Mathew, “Inflammation and pulmonary hypertension,” Cardiology in Review, vol. 18, no. 2, pp. 67–72, 2010. View at Publisher · View at Google Scholar · View at Scopus
- P. Dromparis, G. Sutendra, and E. D. Michelakis, “The role of mitochondria in pulmonary vascular remodeling,” Journal of Molecular Medicine, vol. 88, no. 10, pp. 1003–1010, 2010. View at Publisher · View at Google Scholar · View at Scopus
- P. M. Hassoun, L. Mouthon, J. A. Barberà et al., “Inflammation, growth factors, and pulmonary vascular remodeling,” Journal of the American College of Cardiology, vol. 54, supplement 1, pp. S10–S19, 2009. View at Publisher · View at Google Scholar · View at Scopus
- S. Wedgwood and S. M. Black, “Role of reactive oxygen species in vascular remodeling associated with pulmonary hypertension,” Antioxidants and Redox Signaling, vol. 5, no. 6, pp. 759–769, 2003. View at Google Scholar · View at Scopus
- J. Long, M. J. Russo, C. Muller, and W. T. Vigneswaran, “Surgical treatment of pulmonary hypertension: lung transplantation,” Pulmonary Circulation, vol. 1, pp. 327–333, 2011. View at Google Scholar
- B. S. Goldstein, S. C. Sweet, J. Mao, C. B. Huddleston, and R. M. Grady, “Lung transplantation in children with idiopathic pulmonary arterial hypertension: an 18-year experience,” Journal of Heart and Lung Transplantation, vol. 30, pp. 1148–1152, 2011. View at Publisher · View at Google Scholar · View at Scopus
- E. P. Judge, A. Fabre, H. I. Adamali, and J. J. Egan, “Acute exacerbations and pulmonary hypertension in advanced idiopathic pulmonary fibrosis,” The European Respiratory Journal, vol. 40, no. 1, pp. 93–100, 2012. View at Google Scholar
- V. Hampl, J. Bíbová, A. Baňasová et al., “Pulmonary vascular iNOS induction participates in the onset of chronic hypoxic pulmonary hypertension,” American Journal of Physiology, vol. 290, no. 1, pp. L11–L20, 2006. View at Publisher · View at Google Scholar · View at Scopus
- S. A. Lorch, R. Foust, A. Gow et al., “Immunohistochemical localization of protein 3-nitrotyrosine and S- nitrosocysteine in a murine model of inhaled nitric oxide therapy,” Pediatric Research, vol. 47, no. 6, pp. 798–805, 2000. View at Google Scholar · View at Scopus
- R. Bowers, C. Cool, R. C. Murphy et al., “Oxidative stress in severe pulmonary hypertension,” American Journal of Respiratory and Critical Care Medicine, vol. 169, no. 6, pp. 764–769, 2004. View at Google Scholar · View at Scopus
- R. H. Foxton, J. M. Land, and S. J. R. Heales, “Tetrahydrobiopterin availability in Parkinson's and Alzheimer's disease; potential pathogenic mechanisms,”Neurochemical Research, vol. 32, no. 4-5, pp. 751–756, 2007. View at Publisher ·View at Google Scholar · View at Scopus
- J. S. Beckman and J. P. Crow, “Pathological implications of nitric oxide, superoxide and peroxynitrite formation,” Biochemical Society Transactions, vol. 21, no. 2, pp. 330–334, 1993. View at Google Scholar · View at Scopus
- W. H. Koppenol, J. J. Moreno, W. A. Pryor, H. Ischiropoulos, and J. S. Beckman, “Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide,”Chemical Research in Toxicology, vol. 5, no. 6, pp. 834–842, 1992. View at Publisher· View at Google Scholar · View at Scopus
- Y. Hattori, K. Kasai, and S. S. Gross, “NO suppresses while peroxynitrite sustains NF-κB: a paradigm to rationalize cytoprotective and cytotoxic actions attributed to NO,” Cardiovascular Research, vol. 63, no. 1, pp. 31–40, 2004. View at Publisher ·View at Google Scholar · View at Scopus
- G. Gloire, S. Legrand-Poels, and J. Piette, “NF-κB activation by reactive oxygen species: fifteen years later,” Biochemical Pharmacology, vol. 72, no. 11, pp. 1493–1505, 2006. View at Publisher · View at Google Scholar · View at Scopus
- C. L. M. Cooke and S. T. Davidge, “Peroxynitrite increases iNOS through NF-κB and decreases prostacyclin synthase in endothelial cells,” American Journal of Physiology, vol. 282, no. 2, pp. C395–C402, 2002. View at Google Scholar · View at Scopus
- S. V. Lymar, R. F. Khairutdinov, and J. K. Hurst, “Hydroxyl radical formation by O–O bond homolysis in peroxynitrous acid,” Inorganic Chemistry, vol. 42, no. 17, pp. 5259–5266, 2003. View at Publisher · View at Google Scholar · View at Scopus
- R. Radi, “Nitric oxide, oxidants, and protein tyrosine nitration,” Proceedings of the National Academy of Sciences of the United States of America, vol. 101, no. 12, pp. 4003–4008, 2004. View at Publisher · View at Google Scholar · View at Scopus
- Y. M. W. Janssen-Heininger, M. E. Poynter, and P. A. Baeuerle, “Recent advances towards understanding redox mechanisms in the activation of nuclear factor κB,”Free Radical Biology and Medicine, vol. 28, no. 9, pp. 1317–1327, 2000. View at Publisher · View at Google Scholar · View at Scopus
- R. C. J. Langen, A. M. W. J. Schols, M. C. J. M. Kelders, E. F. M. Wouters, and Y. M. W. Janssen-Heininger, “Inflammatory cytokines inhibit myogenic differentiation through activation of nuclear factor-κB,” The FASEB Journal, vol. 15, no. 7, pp. 1169–1180, 2001. View at Publisher · View at Google Scholar · View at Scopus
- Y. J. Surh, K. S. Chun, H. H. Cha et al., “Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-κB activation,” Mutation Research, vol. 480-481, pp. 243–268, 2001. View at Google Scholar · View at Scopus
- S. C. Chu, J. Marks-Konczalik, H. P. Wu, T. C. Banks, and J. Moss, “Analysis of the cytokine-stimulated human inducible nitric oxide synthase (iNOS) gene: characterization of differences between human and mouse iNOS promoters,”Biochemical and Biophysical Research Communications, vol. 248, no. 3, pp. 871–878, 1998. View at Publisher · View at Google Scholar · View at Scopus
- M. Jaiswal, N. F. LaRusso, L. J. Burgart, and G. J. Gores, “Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism,” Cancer Research, vol. 60, no. 1, pp. 184–190, 2000. View at Google Scholar · View at Scopus
- C. Melchiorri, R. Meliconi, L. Frizziero, et al., “Enhanced and coordinated in vivo expression of inflammatory cytokines and nitric oxide synthase in chondrocytes from patients with osteoarthritis,” Arthritis and Rheumatism, vol. 41, pp. 2165–2174, 1998. View at Google Scholar
- Y. Gutiérrez-Martín, F. J. Martín-Romero, F. Henao, and C. Gutiérrez-Merino, “Synaptosomal plasma membrane Ca2+ pump activity inhibition by repetitive micromolar ONOO− pulses,” Free Radical Biology and Medicine, vol. 32, no. 1, pp. 46–55, 2002. View at Publisher · View at Google Scholar · View at Scopus
- J. Gao, D. Yin, Y. Yao, T. D. Williams, and T. C. Squier, “Progressive decline in the ability of calmodulin isolated from aged brain to activate the plasma membrane Ca-ATPase,” Biochemistry, vol. 37, no. 26, pp. 9536–9548, 1998. View at Publisher ·View at Google Scholar · View at Scopus
- C. R. Hoyal, A. P. Thomas, and H. J. Forman, “Hydroperoxide-induced increases in intracellular calcium due to annexin VI translocation and inactivation of plasma membrane Ca2+-ATPase,” Journal of Biological Chemistry, vol. 271, no. 46, pp. 29205–29210, 1996. View at Publisher · View at Google Scholar · View at Scopus
- D. Yin, K. Kuczera, and T. C. Squier, “The sensitivity of carboxyl-terminal methionines in calmodulin isoforms to oxidation by H2O2 modulates the ability to activate the plasma membrane Ca-ATPase,” Chemical Research in Toxicology, vol. 13, no. 2, pp. 103–110, 2000. View at Publisher · View at Google Scholar · View at Scopus
- R. K. Bartlett, R. J. B. Urbauer, A. Anbanandam, H. S. Smallwood, J. L. Urbauer, and T. C. Squier, “Oxidation of Met144 and Met145 in calmodulin blocks calmodulin dependent activation of the plasma membrane Ca-ATPase,”Biochemistry, vol. 42, no. 11, pp. 3231–3238, 2003. View at Publisher · View at Google Scholar · View at Scopus
- M. P. Kurnellas, A. Nicot, G. E. Shull, and S. Elkabes, “Plasma membrane calcium ATPase deficiency causes neuronal pathology in the spinal cord: a potential mechanism for neurodegeneration in multiple sclerosis and spinal cord injury,”The FASEB Journal, vol. 19, no. 2, pp. 298–300, 2005. View at Publisher · View at Google Scholar · View at Scopus
- B. Khodorov, “Glutamate-induced deregulation of calcium homeostasis and mitochondrial dysfunction in mammalian central neurones,” Progress in Biophysics and Molecular Biology, vol. 86, no. 2, pp. 279–351, 2004. View at Publisher · View at Google Scholar · View at Scopus
- A. C. Rego and C. R. Oliveira, “Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: Implications for the pathogenesis of neurodegenerative diseases,” Neurochemical Research, vol. 28, no. 10, pp. 1563–1574, 2003. View at Publisher · View at Google Scholar · View at Scopus
- J. T. Greenamyre, G. MacKenzie, T. I. Peng, and S. E. Stephans, “Mitochondrial dysfunction in Parkinson's disease,” Biochemical Society Symposium, vol. 66, pp. 85–97, 1999. View at Google Scholar · View at Scopus
- F. Buttgereit and M. D. Brand, “A hierarchy of ATP-consuming processes in mammalian cells,” Biochemical Journal, vol. 312, no. 1, pp. 163–167, 1995. View at Google Scholar · View at Scopus
- P. S. Brookes, Y. Yoon, J. L. Robotham, M. W. Anders, and S. S. Sheu, “Calcium, ATP, and ROS: a mitochondrial love-hate triangle,” American Journal of Physiology, vol. 287, no. 4, pp. C817–C833, 2004. View at Publisher · View at Google Scholar · View at Scopus
- P. Pacher and C. Szabo, “Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease,” American Journal of Pathology, vol. 173, no. 1, pp. 2–13, 2008. View at Publisher · View at Google Scholar · View at Scopus
- C. T. Taylor and S. Moncada, “Nitric oxide, cytochrome C oxidase, and the cellular response to hypoxia,” Arteriosclerosis, Thrombosis, and Vascular Biology, vol. 30, no. 4, pp. 643–647, 2010. View at Publisher · View at Google Scholar · View at Scopus
- J. J. Poderoso, “The formation of peroxynitrite in the applied physiology of mitochondrial nitric oxide,” Archives of Biochemistry and Biophysics, vol. 484, no. 2, pp. 214–220, 2009. View at Publisher · View at Google Scholar · View at Scopus
- P. V. Finocchietto, M. C. Franco, S. Holod et al., “Mitochondrial nitric oxide synthase: a masterpiece of metabolic adaptation, cell growth, transformation, and death,” Experimental Biology and Medicine, vol. 234, no. 9, pp. 1020–1028, 2009.View at Publisher · View at Google Scholar · View at Scopus
- S. Moncada and J. P. Bolaños, “Nitric oxide, cell bioenergetics and neurodegeneration,” Journal of Neurochemistry, vol. 97, no. 6, pp. 1676–1689, 2006. View at Publisher · View at Google Scholar · View at Scopus
- I. Wiswedel, A. Gardemann, A. Storch, D. Peter, and L. Schild, “Degradation of phospholipids by oxidative stress—exceptional significance of cardiolipin,” Free Radical Research, vol. 44, no. 2, pp. 135–145, 2010. View at Publisher · View at Google Scholar · View at Scopus
- H. Bayir, V. A. Tyurin, Y. Y. Tyurina et al., “Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis,” Annals of Neurology, vol. 62, no. 2, pp. 154–169, 2007. View at Publisher · View at Google Scholar · View at Scopus
- S. Pope, J. M. Land, and S. J. R. Heales, “Oxidative stress and mitochondrial dysfunction in neurodegeneration; cardiolipin a critical target?” Biochimica et Biophysica Acta, vol. 1777, no. 7-8, pp. 794–799, 2008. View at Publisher · View at Google Scholar · View at Scopus
- G. Paradies, G. Petrosillo, V. Paradies, and F. M. Ruggiero, “Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease,” Cell Calcium, vol. 45, no. 6, pp. 643–650, 2009. View at Publisher · View at Google Scholar · View at Scopus
- L. A. Macmillan-Crow, J. P. Crow, J. D. Kerby, J. S. Beckman, and J. A. Thompson, “Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts,” Proceedings of the National Academy of Sciences of the United States of America, vol. 93, no. 21, pp. 11853–11858, 1996.View at Publisher · View at Google Scholar · View at Scopus
- L. A. MacMillan-Crow, J. P. Crow, and J. A. Thompson, “Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues,” Biochemistry, vol. 37, no. 6, pp. 1613–1622, 1998.View at Publisher · View at Google Scholar · View at Scopus
- V. Demicheli, C. Quijano, B. Alvarez, and R. Radi, “Inactivation and nitration of human superoxide dismutase (SOD) by fluxes of nitric oxide and superoxide,” Free Radical Biology and Medicine, vol. 42, no. 9, pp. 1359–1368, 2007. View at Publisher · View at Google Scholar · View at Scopus
- E. Cadenas and K. J. A. Davies, “Mitochondrial free radical generation, oxidative stress, and aging,” Free Radical Biology and Medicine, vol. 29, no. 3-4, pp. 222–230, 2000. View at Publisher · View at Google Scholar · View at Scopus
- C. E. Berry and J. M. Hare, “Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications,” Journal of Physiology, vol. 555, pp. 589–606, 2004. View at Publisher · View at Google Scholar· View at Scopus
- G. C. Brown and J. J. Neher, “Inflammatory neurodegeneration and mechanisms of microglial killing of neurons,” Molecular Neurobiology, vol. 41, no. 2-3, pp. 242–247, 2010. View at Publisher · View at Google Scholar · View at Scopus
- A. M. Briones and R. M. Touyz, “Oxidative stress and hypertension: current concepts,” Current Hypertension Reports, vol. 12, no. 2, pp. 135–142, 2010. View at Publisher · View at Google Scholar · View at Scopus
- A. L. Perraud, C. L. Takanishi, B. Shen et al., “Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels,” Journal of Biological Chemistry, vol. 280, no. 7, pp. 6138–6148, 2005.View at Publisher · View at Google Scholar · View at Scopus
- M. Kolisek, A. Beck, A. Fleig, and R. Penner, “Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels,”Molecular Cell, vol. 18, no. 1, pp. 61–69, 2005. View at Publisher · View at Google Scholar · View at Scopus
- B. F. Bessac and S. E. Jordt, “Breathtaking TRP channels: tRPA1 and TRPV1 in airway chemosensation and reflex control,” Physiology, vol. 23, no. 6, pp. 360–370, 2008. View at Publisher · View at Google Scholar · View at Scopus
- M. Trevisani, J. Siemens, S. Materazzi et al., “4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1,” Proceedings of the National Academy of Sciences of the United States of America, vol. 104, no. 33, pp. 13519–13524, 2007. View at Publisher · View at Google Scholar · View at Scopus
- D. A. Andersson, C. Gentry, S. Moss, and S. Bevan, “Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress,” Journal of Neuroscience, vol. 28, no. 10, pp. 2485–2494, 2008. View at Publisher · View at Google Scholar · View at Scopus
- T. Yoshida, R. Inoue, T. Morii et al., “Nitric oxide activates TRP channels by cysteine S-nitrosylation,” Nature Chemical Biology, vol. 2, no. 11, pp. 596–607, 2006. View at Publisher · View at Google Scholar · View at Scopus
- M. L. Pall and J. H. Anderson, “The vanilloid receptor as a putative target of diverse chemicals in multiple chemical sensitivity,” Archives of Environmental Health, vol. 59, no. 7, pp. 363–375, 2004. View at Publisher · View at Google Scholar · View at Scopus
- D. P. Li, S. R. Chen, and H. L. Pan, “VR1 receptor activation induces glutamate release and postsynaptic firing in the paraventricular nucleus,” Journal of Neurophysiology, vol. 92, no. 3, pp. 1807–1816, 2004. View at Publisher · View at Google Scholar · View at Scopus
- C. P. Bailey, K. A. Maubach, and R. S. G. Jones, “Neurokinin-1 receptors in the rat nucleus tractus solitarius: pre- and postsynaptic modulation of glutamate and GABA release,” Neuroscience, vol. 127, no. 2, pp. 467–479, 2004. View at Publisher· View at Google Scholar · View at Scopus
- J. Kamei, H. Tanihara, H. Igarashi, and Y. Kasuya, “Effects of N-methyl-D-aspartate antagonists on the cough reflex,” European Journal of Pharmacology, vol. 168, no. 2, pp. 153–158, 1989. View at Google Scholar · View at Scopus
- T. J. Coderre and R. Melzack, “Central neural mediators of secondary hyperalgesia following heat injury in rats: neuropeptides and excitatory amino acids,”Neuroscience Letters, vol. 131, no. 1, pp. 71–74, 1991. View at Publisher · View at Google Scholar · View at Scopus
- P. M. Dougherty and W. D. Willis, “Enhanced responses of spinothalamic tract neurons to excitatory amino acids accompany capsaicin-induced sensitization in the monkey,” Journal of Neuroscience, vol. 12, no. 3, pp. 883–894, 1992. View at Google Scholar · View at Scopus
- O. K. Andersen, S. Felsby, L. Nicolaisen, P. Bjerring, T. S. Jensen, and L. Arendt-Nielsen, “The effect of Ketamine on stimulation of primary and secondary hyperalgesic areas induced by capsaicin—a double-blind, placebo-controlled, human experimental study,” Pain, vol. 66, no. 1, pp. 51–62, 1996. View at Publisher · View at Google Scholar · View at Scopus
- N. F. Sethna, M. Liu, R. Gracely, G. J. Bennett, and M. B. Max, “Analgesic and cognitive effects of intravenous ketamine-alfentanil combinations versus either drug alone after intradermal capsaicin in normal subjects,” Anesthesia and Analgesia, vol. 86, no. 6, pp. 1250–1256, 1998. View at Publisher · View at Google Scholar · View at Scopus
- D. D. Mitsikostas, M. Sanchez del Rio, C. Waeber, M. A. Moskowitz, and F. M. Cutrer, “The NMDA receptor antagonist MK-801 reduces capsaicin-induced C-fos expression within rat trigeminal nucleus caudalis,” Pain, vol. 76, no. 1-2, pp. 239–248, 1998. View at Publisher · View at Google Scholar · View at Scopus
- T. Kawamata, K. Omote, M. Toriyabe, M. Kawamata, and A. Namiki, “Involvement of capsaicin-sensitive fibers in spinal NMDA-induced glutamate release,” NeuroReport, vol. 12, no. 16, pp. 3447–3450, 2001. View at Google Scholar· View at Scopus
- A. Dray, “Neuropharmacological mechanisms of capsaicin and related substances,”Biochemical Pharmacology, vol. 44, no. 4, pp. 611–615, 1992. View at Publisher ·View at Google Scholar · View at Scopus
- E. Palazzo, V. de Novellis, I. Marabese et al., “Interaction between vanilloid and glutamate receptors in the central modulation of nociception,” European Journal of Pharmacology, vol. 439, no. 1–3, pp. 69–75, 2002. View at Publisher · View at Google Scholar · View at Scopus
- D. K. Lam, B. J. Sessle, B. E. Cairns, and J. W. Hu, “Peripheral NMDA receptor modulation of jaw muscle electromyographic activity induced by capsaicin injection into the temporomandibular joint of rats,” Brain Research, vol. 1046, no. 1-2, pp. 68–76, 2005. View at Publisher · View at Google Scholar · View at Scopus
- C. R. McNamara, J. Mandel-Brehm, D. M. Bautista et al., “TRPA1 mediates formalin-induced pain,” Proceedings of the National Academy of Sciences of the United States of America, vol. 104, no. 33, pp. 13525–13530, 2007. View at Publisher · View at Google Scholar · View at Scopus
- L. H. Piao, T. Fujita, C. Y. Jiang et al., “TRPA1 activation by lidocaine in nerve terminals results in glutamate release increase,” Biochemical and Biophysical Research Communications, vol. 379, no. 4, pp. 980–984, 2009. View at Publisher ·View at Google Scholar · View at Scopus
- M. Kosugi, T. Nakatsuka, T. Fujita, Y. Kuroda, and E. Kumamoto, “Activation of TRPA1 channel facilitates excitatory synaptic transmission in substantia gelatinosa neurons of the adult rat spinal cord,” Journal of Neuroscience, vol. 27, no. 16, pp. 4443–4451, 2007. View at Publisher · View at Google Scholar · View at Scopus
- M. Scott, T. Gomeza, Z. Dinga, and B. Robert, “Differential behavioral effect of the TRPM8/TRPA1 channel agonist icilin (AG-3-5),” European Journal of Pharmacology, vol. 575, no. 1–3, pp. 103–104, 2007. View at Publisher · View at Google Scholar · View at Scopus
- J. Werkheiser, A. Cowan, T. Gomez et al., “Icilin-induced wet-dog shakes in rats are dependent on NMDA receptor activation and nitric oxide production,”Pharmacology Biochemistry and Behavior, vol. 92, no. 3, pp. 543–548, 2009. View at Publisher · View at Google Scholar · View at Scopus
- Z. Ding, A. Cowan, and S. M. Rawls, “Icilin induces a hyperthermia in rats that is dependent on nitric oxide production and NMDA receptor activation,” European Journal of Pharmacology, vol. 578, no. 2-3, pp. 201–208, 2008. View at Publisher ·View at Google Scholar · View at Scopus
- T. Yokoyama, T. Ohbuchi, T. Saito et al., “Allyl isothiocyanates and cinnamaldehyde potentiate miniature excitatory postsynaptic inputs in the supraoptic nucleus in rats,” European Journal of Pharmacology, vol. 655, no. 1–3, pp. 31–37, 2011. View at Publisher · View at Google Scholar · View at Scopus
- C. J. Proudfoot, E. M. Garry, D. F. Cottrell et al., “Analgesia mediated by the TRPM8 cold receptor in chronic neuropathic pain,” Current Biology, vol. 16, no. 16, pp. 1591–1605, 2006. View at Publisher · View at Google Scholar · View at Scopus
- D. Penaloza and J. Arias-Stella, “The heart and pulmonary circulation at high altitudes: healthy highlanders and chronic mountain sickness,” Circulation, vol. 115, no. 9, pp. 1132–1146, 2007. View at Publisher · View at Google Scholar · View at Scopus
- X. Q. Xu and Z. C. Jing, “High-altitude pulmonary hypertension,” European Respiratory Review, vol. 18, pp. 13–17, 2009. View at Google Scholar
- C. V. Remillard and J. X. J. Yuan, “High altitude pulmonary hypertension: role of K+ and Ca2+ channels,” High Altitude Medicine and Biology, vol. 6, no. 2, pp. 133–146, 2005. View at Publisher · View at Google Scholar · View at Scopus
- T. N. Holt and R. J. Callan, “Pulmonary arterial pressure testing for high mountain disease in cattle,” Veterinary Clinics of North America, vol. 23, no. 3, pp. 575–596, 2007. View at Publisher · View at Google Scholar · View at Scopus
- G. L. Colice, N. Hill, Y. J. Lee et al., “Exaggerated pulmonary hypertension with monocrotaline in rats susceptible to chronic mountain sickness,” Journal of Applied Physiology, vol. 83, no. 1, pp. 25–31, 1997. View at Google Scholar · View at Scopus
- J. P. Khoo, L. Zhao, N. J. Alp et al., “Pivotal role for endothelial tetrahydrobiopterin in pulmonary hypertension,” Circulation, vol. 111, no. 16, pp. 2126–2133, 2005.View at Publisher · View at Google Scholar · View at Scopus
- I. Fantozzi, S. Zhang, O. Platoshyn, C. V. Remillard, R. T. Cowling, and J. X. J. Yuan, “Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+entry in human pulmonary artery endothelial cells,” American Journal of Physiology, vol. 285, no. 6, pp. L1233–L1245, 2003. View at Google Scholar · View at Scopus
- P. Bartsch, M. Maggiorini, M. Ritter, C. Noti, P. Vock, and O. Oelz, “Prevention of high-altitude pulmonary edema by nifedipine,” The New England Journal of Medicine, vol. 325, no. 18, pp. 1284–1289, 1991. View at Google Scholar · View at Scopus
- Y. X. Wang, J. Wang, C. Wang et al., “Functional expression of transient receptor potential vanilloid-related channels in chronically hypoxic human pulmonary arterial smooth muscle cells,” Journal of Membrane Biology, vol. 223, no. 3, pp. 151–159, 2008. View at Publisher · View at Google Scholar · View at Scopus
- L. A. Palmer, G. L. Semenza, M. H. Stoler, and R. A. Johns, “Hypoxia induces type II NOS gene expression in pulmonary artery endothelial cells via HIF-1,” American Journal of Physiology, vol. 274, no. 2, pp. L212–L219, 1998. View at Google Scholar· View at Scopus
- A. E. Loot and I. Fleming, “Cytochrome P450-derived epoxyeicosatrienoic acids and pulmonary hypertension: central role of transient receptor potential C6 channels,” Journal of Cardiovascular Pharmacology, vol. 57, no. 2, pp. 140–147, 2011. View at Publisher · View at Google Scholar · View at Scopus
- S. Goerre, M. Wenk, P. Bärtsch et al., “Endothelin-1 in pulmonary hypertension associated with high-altitude exposure,” Circulation, vol. 91, no. 2, pp. 359–364, 1995. View at Google Scholar · View at Scopus
- C. D. Cool, N. F. Voelkel, and T. Bull, “Viral infection and pulmonary hypertension: is there an association?” Expert Review of Respiratory Medicine, vol. 5, no. 2, pp. 207–216, 2011. View at Publisher · View at Google Scholar · View at Scopus
- S. Cicalini, P. Chinello, and N. Petrosillo, “HIV infection and pulmonary arterial hypertension,” Expert Review of Respiratory Medicine, vol. 5, no. 2, pp. 257–266, 2011. View at Publisher · View at Google Scholar · View at Scopus
- P. Y. Hsue, S. G. Deeks, H. H. Farah et al., “Role of HIV and human herpesvirus-8 infection in pulmonary arterial hypertension,” AIDS, vol. 22, no. 7, pp. 825–833, 2008. View at Publisher · View at Google Scholar · View at Scopus
- T. M. Bull, C. A. Meadows, C. D. Coldren et al., “Human herpesvirus-8 infection of primary pulmonary microvascular endothelial cells,” American Journal of Respiratory Cell and Molecular Biology, vol. 39, no. 6, pp. 706–716, 2008. View at Publisher · View at Google Scholar · View at Scopus
- S. Janda, B. S. Quon, and J. Swiston, “HIV and pulmonary arterial hypertension: a systematic review,” HIV Medicine, vol. 11, no. 10, pp. 620–634, 2010. View at Publisher · View at Google Scholar · View at Scopus
- A. Talwar, P. Sarkar, and M. J. Rosen, “Pulmonary arterial hypertension in human immunodeficiency virus infection,” Postgraduate Medicine, vol. 121, no. 5, pp. 56–67, 2009. View at Publisher · View at Google Scholar · View at Scopus
- A. Cota-Gomez, A. C. Flores, X.-F. Ling, M. Varella-Garcia, and S. C. Flores, “HIV-1 Tat increases oxidant burden in the lungs of transgenic mice,” Free Radical Biology and Medicine, vol. 51, no. 9, pp. 1697–1707, 2011. View at Publisher · View at Google Scholar · View at Scopus
- C. Tcherakian, É. Rivaud, É. Catherinot, D. Zucman, A.-C. Metivier, and L.-J. Couderc, “Pulmonary arterial hypertension related to HIV: is inflammation related to IL-6 the cornerstone?” Revue de Pneumologie Clinique, vol. 67, no. 4, pp. 250–257, 2011. View at Publisher · View at Google Scholar · View at Scopus
- J.-L. Lü, J. Nurko, J. Jiang et al., “Nordihydroguaiaretic acid (NDGA) inhibits ritonavir-induced endothelial dysfunction in porcine pulmonary arteries,” Medical Science Monitor, vol. 17, no. 11, pp. BR312–BR318, 2011. View at Google Scholar ·View at Scopus
- S. M. Weakley, J. Jiang, J. Lü et al., “Natural antioxidant dihydroxybenzyl alcohol blocks ritonavir-induced endothelial dysfunction in porcine pulmonary arteries and human endothelial cells,” Medical Science Monitor, vol. 17, no. 9, pp. BR235–BR241, 2011. View at Google Scholar · View at Scopus
- C. Cheng, X. Wang, S. M. Weakley et al., “The soybean isoflavonoid equol blocks ritonavir-induced endothelial dysfunction in porcine pulmonary arteries and human pulmonary artery endothelial cells,” Journal of Nutrition, vol. 140, no. 1, pp. 12–17, 2010. View at Publisher · View at Google Scholar · View at Scopus
- H. Chai, S. Yan, P. Lin, A. B. Lumsden, Q. Yao, and C. Chen, “Curcumin blocks HIV protease inhibitor ritonavir-induced vascular dysfunction in porcine coronary arteries,” Journal of the American College of Surgeons, vol. 200, no. 6, pp. 820–830, 2005. View at Publisher · View at Google Scholar · View at Scopus
- W. Deng, L. Baki, J. Yin, H. Zhou, and C. M. Baumgarten, “HIV protease inhibitors elicit volume-sensitive Cl− current in cardiac myocytes via mitochondrial ROS,” Journal of Molecular and Cellular Cardiology, vol. 49, no. 5, pp. 746–752, 2010. View at Publisher · View at Google Scholar · View at Scopus
- Z. Vyslouzil, J. Polak, J. Widimsky, and M. Sukova, “Pathogenesis of pulmonary hypertension in tuberculosis,” Czechoslovak Medicine, vol. 3, no. 2, pp. 123–131, 1980. View at Google Scholar · View at Scopus
- A. E. H. Ahmed, A. S. Ibrahim, and S. M. Elshafie, “Pulmonary hypertension in patients with treated pulmonary tuberculosis: analysis of 14 consecutive cases,”Clinical Medicine Insights, vol. 5, no. 1, pp. 1–5, 2011. View at Publisher · View at Google Scholar · View at Scopus
- K. Machida and R. Maekura, “State of the art: sequelae of tuberculosis,” Kekkaku, vol. 80, no. 10, pp. 655–674, 2005. View at Google Scholar · View at Scopus
- E. D. Chan, K. R. Morris, J. T. Belisle et al., “Induction of inducible nitric oxide synthase-NO* by lipoarabinomannan of Mycobacterium tuberculosis is mediated by MEK1-ERK, MKK7-JNK, and NF-κB signaling pathways,” Infection and Immunity, vol. 69, no. 4, pp. 2001–2010, 2001. View at Publisher · View at Google Scholar · View at Scopus
- A. M. Siore, R. E. Parker, A. A. Stecenko et al., “Endotoxin-induced acute lung injury requires interaction with the liver,” American Journal of Physiology, vol. 289, no. 5, pp. L769–L776, 2005. View at Publisher · View at Google Scholar · View at Scopus
- M. Lipcsey, E. Söderberg, S. Basu et al., “F-2-isoprostane, inflammation, cardiac function and oxygenation in the endotoxaemic pig,” Prostaglandins Leukotrienes and Essential Fatty Acids, vol. 78, no. 3, pp. 209–217, 2008. View at Publisher ·View at Google Scholar · View at Scopus
- P. K. Gonzalez, J. Zhuang, S. R. Doctrow et al., “Role of oxidant stress in the adult respiratory distress syndrome: evaluation of a novel antioxidant strategy in a porcine model of endotoxin-induced acute lung injury,” Shock, vol. 6, supplement 6, pp. S23–S26, 1996. View at Google Scholar · View at Scopus
- R. H. Demling, M. Smith, R. Gunther, and T. Wandzilak, “Endotoxin-induced lung injury in unanesthetized sheep: effect of methylprednisolone,” Circulatory Shock, vol. 8, no. 3, pp. 351–360, 1981. View at Google Scholar · View at Scopus
- C. Boer, A. B. J. Groeneveld, G. J. Scheffer, J. J. De Lange, N. Westerhof, and P. Sipkema, “Induced nitric oxide impairs relaxation but not contraction in endotoxin-exposed rat pulmonary arteries,” Journal of Surgical Research, vol. 127, no. 2, pp. 197–202, 2005. View at Publisher · View at Google Scholar · View at Scopus
- R. Rodrigo, O. Cauli, J. Boix, N. ElMlili, A. Agusti, and V. Felipo, “Role of NMDA receptors in acute liver failure and ammonia toxicity: therapeutical implications,”Neurochemistry International, vol. 55, no. 1–3, pp. 113–118, 2009. View at Publisher · View at Google Scholar · View at Scopus
- P. Monfort, E. Kosenko, S. Erceg, J. J. Canales, and V. Felipo, “Molecular mechanism of acute ammonia toxicity: role of NMDA receptors,” Neurochemistry International, vol. 41, no. 2-3, pp. 95–102, 2002. View at Publisher · View at Google Scholar · View at Scopus
- C. Rose, “Increased extracellular brain glutamate in acute liver failure: decreased uptake or increased release?” Metabolic Brain Disease, vol. 17, no. 4, pp. 251–261, 2002. View at Publisher · View at Google Scholar · View at Scopus
- P. Gustin, B. Urbain, J. F. Prouvost, and M. Ansay, “Effects of atmospheric ammonia on pulmonary hemodynamics and vascular permeability in pigs: interaction with endotoxins,” Toxicology and Applied Pharmacology, vol. 125, no. 1, pp. 17–26, 1994. View at Publisher · View at Google Scholar · View at Scopus
- J. C. Leung, B. R. Travis, J. W. Verlander et al., “Expression and developmental regulation of the NMDA receptor subunits in the kidney and cardiovascular system,” American Journal of Physiology, vol. 283, no. 4, pp. R964–R971, 2002.View at Google Scholar · View at Scopus
- T. Nassar, K. Bdeir, S. Yarovoi et al., “tPA regulates pulmonary vascular activity through NMDA receptors,” American Journal of Physiology, vol. 301, no. 3, pp. L307–L314, 2011. View at Publisher · View at Google Scholar · View at Scopus
- R. Ben-Abraham, M. Guttman, R. Flaishon, N. Marouani, D. Niv, and A. A. Weinbroum, “Mesenteric artery clamping/unclamping-induced acute lung injury is attenuated by N-methyl-D-aspartate antagonist dextromethorphan,” Lung, vol. 184, no. 6, pp. 309–317, 2006. View at Publisher · View at Google Scholar · View at Scopus
- A. C. Arroliga, S. Sandur, D. W. Jacobsen et al., “Association between hyperhomocysteinemia and primary pulmonary hypertension,” Respiratory Medicine, vol. 97, no. 7, pp. 825–829, 2003. View at Publisher · View at Google Scholar · View at Scopus
- I. H. Ozerol, F. A. Pac, E. Ozerol et al., “Plasma endothelin-1, homocysteine and serum nitric oxide values in patients with left-to-right shunt,” Indian Heart Journal, vol. 56, no. 6, pp. 653–657, 2004. View at Google Scholar · View at Scopus
- S. Szamosi, Z. Csiki, E. Szomják et al., “Plasma homocysteine levels, the prevalence of methylenetetrahydrofolate reductase gene C677T polymorphism and macrovascular disorders in systemic sclerosis: risk factors for accelerated macrovascular damage?” Clinical Reviews in Allergy and Immunology, vol. 36, no. 2-3, pp. 145–149, 2009. View at Publisher · View at Google Scholar · View at Scopus
- P. H. Rolland, A. Friggi, A. Barlatier et al., “Hyperhomocysteinemia-induced vascular damage in the minipig: captopril-hydrochlorothiazide combination prevents elastic alterations,” Circulation, vol. 91, no. 4, pp. 1161–1174, 1995. View at Google Scholar · View at Scopus
- L. B. R. Zocrato, L. S. A. Capettini, B. A. Rezende et al., “Increased expression of endothelial iNOS accounts for hyporesponsiveness of pulmonary artery to vasoconstrictors after paraquat poisoning,” Toxicology in Vitro, vol. 24, no. 3, pp. 1019–1025, 2010. View at Publisher · View at Google Scholar · View at Scopus
- M. Singh, V. Murthy, and C. Ramassamy, “Standardized extracts of Bacopa monniera protects against MPP+ and paraquat induce-toxicities by modulation mitochondrial activities, proteasomal functions and redox pathways,” Toxicological Sciences, vol. 125, no. 1, pp. 219–232, 2012. View at Google Scholar
- A. Czerniczyniec, A. G. Karadayian, J. Bustamante, R. A. Cutrera, and S. Lores-Arnaiz, “Paraquat induces behavioral changes and cortical and striatal mitochondrial dysfunction,” Free Radical Biology and Medicine, vol. 51, pp. 1428–1436, 2011. View at Publisher · View at Google Scholar · View at Scopus
- K. Shimizu, K. Matsubara, K. I. Ohtaki, and H. Shiono, “Paraquat leads to dopaminergic neural vulnerability in organotypic midbrain culture,” Neuroscience Research, vol. 46, no. 4, pp. 523–532, 2003. View at Publisher · View at Google Scholar · View at Scopus
- E. N. Mangano, D. Litteljohn, R. So et al., “Interferon-γ plays a role in paraquat-induced neurodegeneration involving oxidative and proinflammatory pathways,”Neurobiology of Aging, vol. 33, no. 7, pp. 1411–1426, 2011. View at Publisher · View at Google Scholar · View at Scopus
- A. F. Fernandes, J. Zhou, X. Zhang et al., “Oxidative inactivation of the proteasome in retinal pigment epithelial cells: a potential link between oxidative stress and up-regulation of interleukin-8,” Journal of Biological Chemistry, vol. 283, no. 30, pp. 20745–20753, 2008. View at Publisher · View at Google Scholar · View at Scopus
- S. L. Chow, V. Chandran, R. Fazelzad, and S. R. Johnson, “Prognostic factors for survival in systemic lupus erythematosus associated pulmonary hypertension,”Lupus, vol. 21, no. 4, pp. 353–364, 2012. View at Publisher · View at Google Scholar· View at Scopus
- J. G. Coghlan, J. Pope, and C. P. Denton, “Assessment of endpoints in pulmonary arterial hypertension associated with connective tissue disease,” Current Opinion in Pulmonary Medicine, vol. 16, supplement 1, pp. S27–S34, 2010. View at Publisher ·View at Google Scholar · View at Scopus
- I. Koniari, S. N. Siminelakis, N. G. Baikoussis, G. Papadopoulos, J. Goudevenos, and E. Apostolakis, “Antiphospholipid syndrome; its implication in cardiovascular diseases: a review,” Journal of Cardiothoracic Surgery, vol. 5, no. 1, article 101, 2010. View at Publisher · View at Google Scholar · View at Scopus
- S. R. Johnson and J. T. Granton, “Pulmonary hypertension in systemic sclerosis and systemic lupus erythematosus,” European Respiratory Review, vol. 20, no. 122, pp. 277–286, 2011. View at Publisher · View at Google Scholar · View at Scopus
- R. Ahmad, Z. Rasheed, and H. Ahsan, “Biochemical and cellular toxicology of peroxynitrite: implications in cell death and autoimmune phenomenon,”Immunopharmacology and Immunotoxicology, vol. 31, no. 3, pp. 388–396, 2009.View at Publisher · View at Google Scholar · View at Scopus
- M. Humbert, “Pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: pathophysiology,” European Respiratory Review, vol. 19, no. 115, pp. 59–63, 2010. View at Publisher · View at Google Scholar · View at Scopus
- K. L. Lane, M. Talati, E. Austin, et al., “Oxidative injury is a common consequence of BMPR2 mutations,” Pulmonary Circulation, vol. 1, pp. 72–83, 2011. View at Google Scholar
- M. D. Bear, M. Li, Y. Liu, M. A. Giel-Moloney, B. L. Fanburg, and D. Toksoz, “The Lbc Rho guanine nucleotide exchange factor/α-catulin axis functions in serotonin-induced vascular smooth muscle cell mitogenesis and RhoA/ROCK activation,”Journal of Biological Chemistry, vol. 285, no. 43, pp. 32919–32926, 2010. View at Publisher · View at Google Scholar · View at Scopus
- X. Y. Chen, J. N. Dun, Q. F. Miao, and Y. J. Zhang, “Fasudil hydrochloride hydrate, a Rho-kinase inhibitor, suppresses 5-hydroxytryptamine-induced pulmonary artery smooth muscle cell proliferation via JNK and ERK1/2 pathway,”Pharmacology, vol. 83, no. 2, pp. 67–79, 2009. View at Publisher · View at Google Scholar · View at Scopus
- D. A. Zopf, L . A. D. Neves, K. J. Nikula, J. Huang, P. B. Senese, and M. R. Gralinski, “C-122, a novel antagonist of serotonin receptor 5-HT2B, prevents monocrotaline-induced pulmonary arterial hypertension in rats,” European Journal of Pharmacology, vol. 670, no. 1, pp. 195–203, 2011. View at Publisher ·View at Google Scholar · View at Scopus
- M. R. MacLean and Y. Dempsie, “The serotonin hypothesis of pulmonary hypertension revisited,” Advances in Experimental Medicine and Biology, vol. 661, pp. 309–322, 2010. View at Publisher · View at Google Scholar · View at Scopus
- Y. Dempsie, I. Morecroft, D. J. Welsh et al., “Converging evidence in support of the serotonin hypothesis of dexfenfluramine-induced pulmonary hypertension with novel transgenic mice,” Circulation, vol. 117, no. 22, pp. 2928–2937, 2008. View at Publisher · View at Google Scholar · View at Scopus
- C. M. Villalón and D. Centurión, “Cardiovascular responses produced by 5-hydroxytriptamine:a pharmacological update on the receptors/mechanisms involved and therapeutic implications,” Naunyn-Schmiedeberg's Archives of Pharmacology, vol. 376, no. 1-2, pp. 45–63, 2007. View at Google Scholar · View at Scopus
- N. Desbuards, D. Antier, G. Y. Rochefort et al., “Dexfenfluramine discontinuous treatment does not worsen hypoxia-induced pulmonary vascular remodeling but activates RhoA/ROCK pathway: consequences on pulmonary hypertension,”European Journal of Pharmacology, vol. 602, no. 2-3, pp. 355–363, 2009. View at Publisher · View at Google Scholar · View at Scopus
- A. R. Hemnes, A. Zaiman, and H. C. Champion, “PDE5A inhibition attenuates bleomycin-induced pulmonary fibrosis and pulmonary hypertension through inhibition of ROS generation and RhoA/Rho kinase activation,” American Journal of Physiology, vol. 294, no. 1, pp. L24–L33, 2008. View at Publisher · View at Google Scholar · View at Scopus
- L. C. Price, D. Montani, C. Tcherakian et al., “Dexamethasone reverses monocrotaline-induced pulmonary arterial hypertension in rats,” European Respiratory Journal, vol. 37, no. 4, pp. 813–822, 2011. View at Publisher · View at Google Scholar · View at Scopus
- M. Oka, N. Homma, and I. F. McMurtry, “Rho kinase-mediated vasoconstriction in rat models of pulmonary hypertension,” Methods in Enzymology, vol. 439, pp. 191–204, 2008. View at Publisher · View at Google Scholar · View at Scopus
- I. F. McMurtry, K. Abe, H. Ota, K. A. Fagan, and M. Oka, “Rho kinase-mediated vasoconstriction in pulmonary hypertension,” Advances in Experimental Medicine and Biology, vol. 661, pp. 299–308, 2010. View at Publisher · View at Google Scholar · View at Scopus
- M. J. Connolly and P. I. Aaronson, “Key role of the RhoA/Rho kinase system in pulmonary hypertension,” Pulmonary Pharmacology and Therapeutics, vol. 24, no. 1, pp. 1–14, 2011. View at Publisher · View at Google Scholar · View at Scopus
- M. Thomas, “Pharmacological targets for pulmonary vascular disease: vasodilation versus anti-remodelling,” Advances in Experimental Medicine and Biology, vol. 661, pp. 475–490, 2010. View at Publisher · View at Google Scholar · View at Scopus
- S. Shimizu, M. Tahara, S. Ogata et al., “Involvement of nuclear factor-kB activation through RhoA/Rho-kinase pathway in LPS-induced IL-8 production in human cervical stromal cells,” Molecular Human Reproduction, vol. 13, no. 3, pp. 181–187, 2007. View at Publisher · View at Google Scholar · View at Scopus
- K. Goto, Y. Chiba, K. Matsusue et al., “The proximal STAT6 and NF-κB sites are responsible for IL-13- and TNF-α-induced RhoA transcriptions in human bronchial smooth muscle cells,” Pharmacological Research, vol. 61, no. 5, pp. 466–472, 2010. View at Publisher · View at Google Scholar · View at Scopus
- K. Goto, Y. Chiba, H. Sakai, and M. Misawa, “Mechanism of inhibitory effect of prednisolone on RhoA upregulation in human bronchial smooth muscle cells,”Biological and Pharmaceutical Bulletin, vol. 33, no. 4, pp. 710–713, 2010. View at Publisher · View at Google Scholar · View at Scopus
- E. Kakiashvili, Q. Dan, M. Vandermeer et al., “The epidermal growth factor receptor mediates tumor necrosis factor-α-induced activation of the ERK/GEF-H1/RhoA pathway in tubular epithelium,” Journal of Biological Chemistry, vol. 286, no. 11, pp. 9268–9279, 2011. View at Publisher · View at Google Scholar · View at Scopus
- J. Peng, F. He, C. Zhang, X. Deng, and F. Yin, “Protein kinase C-α signals P115RhoGEF phosphorylation and RhoA activation in TNF-α-induced mouse brain microvascular endothelial cell barrier dysfunction,” Journal of Neuroinflammation, vol. 8, article 28, 2011. View at Publisher · View at Google Scholar · View at Scopus
- T. Nakakuki, M. Ito, H. Iwasaki et al., “Rho/Rho-kinase pathway contributes to C-reactive protein-induced plasminogen activator inhibitor-1 expression in endothelial cells,” Arteriosclerosis, Thrombosis, and Vascular Biology, vol. 25, no. 10, pp. 2088–2093, 2005. View at Publisher · View at Google Scholar · View at Scopus
- L. Jin, Z. Ying, and R. C. Webb, “Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta,” American Journal of Physiology, vol. 287, no. 4, pp. H1495–H1500, 2004. View at Publisher · View at Google Scholar · View at Scopus
- S. Ryoo, A. Bhunia, F. Chang, A. Shoukas, D. E. Berkowitz, and L. H. Romer, “OxLDL-dependent activation of arginase II is dependent on the LOX-1 receptor and downstream RhoA signaling,” Atherosclerosis, vol. 214, no. 2, pp. 279–287, 2011. View at Publisher · View at Google Scholar · View at Scopus
- S. Chandra, M. J. Romero, A. Shatanawi, A. M. Alkilany, R. B. Caldwell, and R. W. Caldwell, “Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway,” British Journal of Pharmacology, vol. 165, no. 2, pp. 506–519, 2012. View at Publisher · View at Google Scholar · View at Scopus
- T. C. Resta, B. R. S. Broughton, and N. L. Jernigan, “Reactive oxygen species and RhoA signaling in vascular smooth muscle: role in chronic hypoxia-induced pulmonary hypertension,” Advances in Experimental Medicine and Biology, vol. 661, pp. 355–373, 2010. View at Publisher · View at Google Scholar · View at Scopus
- B. R. S. Broughton, N. L. Jernigan, C. E. Norton, B. R. Walker, and T. C. Resta, “Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA,” American Journal of Physiology, vol. 298, no. 2, pp. L232–L242, 2010. View at Publisher · View at Google Scholar · View at Scopus
- J. Ren, J. Duan, D. P. Thomas et al., “IGF-I alleviates diabetes-induced RhoA activation, eNOS uncoupling, and myocardial dysfunction,” American Journal of Physiology, vol. 294, no. 3, pp. R793–R802, 2008. View at Publisher · View at Google Scholar · View at Scopus
- L. Yao, M. J. Romero, H. A. Toque, G. Yang, R. B. Caldwell, and R. W. Caldwell, “The role of RhoA/Rho kinase pathway in endothelial dysfunction,” Journal of Cardiovascular Disease Research, vol. 1, no. 4, pp. 165–170, 2010. View at Publisher· View at Google Scholar · View at Scopus
- L. Zhao, X. Wang, Q. Chang et al., “Neferine, a bisbenzylisoquinline alkaloid attenuates bleomycin-induced pulmonary fibrosis,” European Journal of Pharmacology, vol. 627, no. 1–3, pp. 304–312, 2010. View at Publisher · View at Google Scholar · View at Scopus
- F. Dong, S. Soubeyrand, and R. J. G. Haché, “Activation of PARP-1 in response to bleomycin depends on the Ku antigen and protein phosphatase 5,” Oncogene, vol. 29, no. 14, pp. 2093–2103, 2010. View at Publisher · View at Google Scholar · View at Scopus
- O. Mungunsukh, A. J. Griffin, Y. H. Lee, and R. M. Day, “Bleomycin induces the extrinsic apoptotic pathway in pulmonary endothelial cells,” American Journal of Physiology, vol. 298, no. 5, pp. L696–L703, 2010. View at Publisher · View at Google Scholar · View at Scopus
- V. G. Desai, A. Aidoo, J. Li, L. E. Lyn-Cook, D. A. Casciano, and R. J. Feuers, “Effects of bleomycin on liver antioxidant enzymes and the electron transport system from ad libitum-fed and dietary-restricted female and male Fischer 344 rats,” Nutrition and Cancer, vol. 36, no. 1, pp. 42–51, 2000. View at Google Scholar· View at Scopus
- Z. Van Rheen, C. Fattman, S. Domarski et al., “Lung extracellular superoxide dismutase overexpression lessens bleomycin-induced pulmonary hypertension and vascular remodeling,” American Journal of Respiratory Cell and Molecular Biology, vol. 44, no. 4, pp. 500–508, 2011. View at Publisher · View at Google Scholar · View at Scopus
- S. I. Said, S. A. Hamidi, K. G. Dickman et al., “Moderate pulmonary arterial hypertension in male mice lacking the vasoactive intestinal peptide gene,”Circulation, vol. 115, no. 10, pp. 1260–1268, 2007. View at Publisher · View at Google Scholar · View at Scopus
- S. A. Hamidi, S. Prabhakar, and S. I. Said, “Enhancement of pulmonary vascular remodelling and inflammatory genes with VIP gene deletion,” European Respiratory Journal, vol. 31, no. 1, pp. 135–139, 2008. View at Publisher · View at Google Scholar · View at Scopus
- S. A. Hamidi, R. Z. Lin, A. M. Szema, S. Lyubsky, Y. P. Jiang, and S. I. Said, “VIP and endothelin receptor antagonist: an effective combination against experimental pulmonary arterial hypertension,” Respiratory Research, vol. 12, article 141, 2011.View at Publisher · View at Google Scholar · View at Scopus
- P. Z. Anastasiadis, L. Bezin, L. J. Gordon, B. Imerman, J. Blitz, and R. A. Levine, “Vasoactive intestinal peptide induces both tyrosine hydroxylase activity and tetrahydrobiopterin biosynthesis in PC12 cells,” Neuroscience, vol. 86, no. 1, pp. 179–189, 1998. View at Publisher · View at Google Scholar · View at Scopus
- X. Z. Shi and S. K. Sarna, “Homeostatic and therapeutic roles of VIP in smooth muscle function: Myo-neuroimmune interactions,” American Journal of Physiology, vol. 297, no. 4, pp. G716–G725, 2009. View at Publisher · View at Google Scholar · View at Scopus
- R. Yu, H. Zhang, L. Huang, X. Liu, and J. Chen, “Anti-hyperglycemic, antioxidant and anti-inflammatory effects of VIP and a VPAC1 agonist on streptozotocin-induced diabetic mice,” Peptides, vol. 32, no. 2, pp. 216–222, 2011. View at Publisher · View at Google Scholar · View at Scopus
- M. Nandi, A. Miller, R. Stidwill et al., “Pulmonary hypertension in a GTP-cyclohydrolase 1-deficient mouse,” Circulation, vol. 111, no. 16, pp. 2086–2090, 2005. View at Publisher · View at Google Scholar · View at Scopus
- J. Belik, B. A. S. McIntyre, M. Enomoto, J. Pan, H. Grasemann, and J. Vasquez-Vivar, “Pulmonary hypertension in the newborn GTP cyclohydrolase I-deficient mouse,” Free Radical Biology and Medicine, vol. 51, no. 12, pp. 2227–2233, 2011.View at Publisher · View at Google Scholar · View at Scopus
- L. V. d'Uscio, “ENOS uncoupling in pulmonary hypertension,” Cardiovascular Research, vol. 92, no. 3, pp. 359–360, 2011. View at Publisher · View at Google Scholar · View at Scopus
- M. Yanagisawa, H. Kurihara, S. Kimura et al., “A novel potent vasoconstrictor peptide produced by vascular endothelial cells,” Nature, vol. 332, no. 6163, pp. 411–415, 1988. View at Google Scholar · View at Scopus
- M. La and J. J. Reid, “Endothelin-1 and the regulation of vascular tone,” Clinical and Experimental Pharmacology and Physiology, vol. 22, no. 5, pp. 315–323, 1995.View at Google Scholar · View at Scopus
- A. Giaid, M. Yanagisawa, D. Langleben et al., “Expression of endothelin-1 in the lungs of patients with pulmonary hypertension,” The New England Journal of Medicine, vol. 328, no. 24, pp. 1732–1739, 1993. View at Publisher · View at Google Scholar · View at Scopus
- R. N. Channick, G. Simonneau, O. Sitbon et al., “Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study,” The Lancet, vol. 358, no. 9288, pp. 1119–1123, 2001. View at Publisher · View at Google Scholar · View at Scopus
- M. Woods, D. Bishop-Bailey, J. R. Pepper, T. W. Evans, J. A. Mitchell, and T. D. Warner, “Cytokine and lipopolysaccharide stimulation of endothelin-1 release from human internal mammary artery and saphenous vein smooth-muscle cells,”Journal of Cardiovascular Pharmacology, vol. 31, supplement 1, pp. S348–S350, 1998. View at Google Scholar · View at Scopus
- M. Woods, J. A. Mitchell, E. G. Wood et al., “Endothelin-1 is induced by cytokines in human vascular smooth muscle cells: evidence for intracellular endothelin-converting enzyme,” Molecular Pharmacology, vol. 55, no. 5, pp. 902–909, 1999.View at Google Scholar · View at Scopus
- S. Narayan, G. Prasanna, R. R. Krishnamoorthy, X. Zhang, and T. Yorio, “Endothelin-1 synthesis and secretion in human retinal pigment epithelial cells (ARPE-19): differential regulation by cholinergics and TNF-alpha,” Investigative Ophthalmology and Visual Science, vol. 44, no. 11, pp. 4885–4894, 2003. View at Publisher · View at Google Scholar · View at Scopus
- J. Ruef, M. Moser, W. Kübler, and C. Bode, “Induction of endothelin-1 expression by oxidative stress in vascular smooth muscle cells,” Cardiovascular Pathology, vol. 10, no. 6, pp. 311–315, 2001. View at Publisher · View at Google Scholar · View at Scopus
- S. J. An, R. Boyd, M. Zhu, A. Chapman, D. R. Pimentel, and H. D. Wang, “NADPH oxidase mediates angiotensin II-induced endothelin-1 expression in vascular adventitial fibroblasts,” Cardiovascular Research, vol. 75, no. 4, pp. 702–709, 2007.View at Publisher · View at Google Scholar · View at Scopus
- R. Jiménez, R. López-Sepúlveda, M. Kadmiri et al., “Polyphenols restore endothelial function in DOCA-salt hypertension: role of endothelin-1 and NADPH oxidase,” Free Radical Biology and Medicine, vol. 43, no. 3, pp. 462–473, 2007. View at Publisher · View at Google Scholar · View at Scopus
- M. Woods, E. G. Wood, S. C. Bardswell et al., “Role for nuclear factor-κB and signal transducer and activator of transcription 1/interferon regulatory factor-1 in cytokine-induced endothelin-1 release in human vascular smooth muscle cells,”Molecular Pharmacology, vol. 64, no. 4, pp. 923–931, 2003. View at Publisher ·View at Google Scholar · View at Scopus
- P. Henno, C. Maurey, C. Danel et al., “Pulmonary vascular dysfunction in endstage cystic fibrosis: role of NF-κB and endothelin-1,” European Respiratory Journal, vol. 34, no. 6, pp. 1329–1337, 2009. View at Publisher · View at Google Scholar · View at Scopus
- D. N. Müller, A. Fiebeler, J. K. Park, R. Dechend, and F. C. Luft, “Angiotensin II and endothelin induce inflammation and thereby promote hypertension-induced end-organ damage,” Clinical Nephrology, vol. 60, supplement 1, pp. S2–S12, 2003.View at Google Scholar · View at Scopus
- D. Ramzy, V. Rao, L. C. Tumiati et al., “Tetrahydrobiopterin prevents cyclosporine-induced vasomotor dysfunction,” Transplantation, vol. 79, no. 8, pp. 876–881, 2005. View at Publisher · View at Google Scholar · View at Scopus
- V. A. Ohanyan, G. Guarini, C. K. Thodeti et al., “Endothelin-mediated in vivo pressor responses following TRPV1 activation,” American Journal of Physiology, vol. 301, no. 3, pp. H1135–H1142, 2011. View at Publisher · View at Google Scholar · View at Scopus
- C. Xie and D. H. Wang, “Ablation of transient receptor potential vanilloid 1 abolishes endothelin-induced increases in afferent renal nerve activity: mechanisms and functional significance,” Hypertension, vol. 54, no. 6, pp. 1298–1305, 2009. View at Publisher · View at Google Scholar · View at Scopus
- H. B. He, D. Z. Dai, and Y. Dai, “CPU0213, a novel endothelin receptor antagonist, ameliorates septic renal lesion by suppressing ET system and NF-κB in rats,” Acta Pharmacologica Sinica, vol. 27, no. 9, pp. 1213–1221, 2006. View at Publisher ·View at Google Scholar · View at Scopus
- S. Wedgwood, D. M. McMullan, J. M. Bekker, J. R. Fineman, and S. M. Black, “Role for endothelin-1-induced superoxide and peroxynitrite production in rebound pulmonary hypertension associated with inhaled nitric oxide therapy,”Circulation Research, vol. 89, no. 4, pp. 357–364, 2001. View at Google Scholar ·View at Scopus
- M. Romero, R. Jiménez, M. Sánchez et al., “Quercetin inhibits vascular superoxide production induced by endothelin-1: role of NADPH oxidase, uncoupled eNOS and PKC,” Atherosclerosis, vol. 202, no. 1, pp. 58–67, 2009. View at Publisher ·View at Google Scholar · View at Scopus
- E. D. Loomis, J. C. Sullivan, D. A. Osmond, D. M. Pollock, and J. S. Pollock, “Endothelin mediates superoxide production and vasoconstriction through activation of NADPH oxidase and uncoupled nitric-oxide synthase in the rat aorta,” Journal of Pharmacology and Experimental Therapeutics, vol. 315, no. 3, pp. 1058–1064, 2005. View at Publisher · View at Google Scholar · View at Scopus
- J. S. Zheng, X. Q. Yang, K. J. Lookingland et al., “Gene transfer of human guanosine 5′-triphosphate cyclohydrolase I restores vascular tetrahydrobiopterin level and endothelial function in low renin hypertension,” Circulation, vol. 108, no. 10, pp. 1238–1245, 2003. View at Publisher · View at Google Scholar · View at Scopus
- R. H. Steinhorn, J. A. Russell, S. Lakshminrusimha, S. F. Gugino, S. M. Black, and J. R. Fineman, “Altered endothelium-dependent relaxations in lambs with high pulmonary blood flow and pulmonary hypertension,” American Journal of Physiology, vol. 280, no. 1, pp. H311–H317, 2001. View at Google Scholar · View at Scopus
- H. J. Xia, D. Z. Dai, and Y. Dai, “Up-regulated inflammatory factors endothelin, NFκB, TNFα and iNOS involved in exaggerated cardiac arrhythmias in l-thyroxine-induced cardiomyopathy are suppressed by darusentan in rats,” Life Sciences, vol. 79, no. 19, pp. 1812–1819, 2006. View at Publisher · View at Google Scholar · View at Scopus
- K. Sato, D. M. Rodman, and I. F. McMurtry, “Hypoxia inhibits increased ET(B) receptor-mediated NO synthesis in hypertensive rat lungs,” American Journal of Physiology, vol. 276, no. 4, pp. L571–L581, 1999. View at Google Scholar · View at Scopus
- J. Hirahashi, T. Nakaki, K. Hishikawa et al., “Endothelin-1 inhibits induction of nitric oxide synthase and GTP cyclohydrolase I in rat mesangial cells,”Pharmacology, vol. 53, no. 4, pp. 241–249, 1996. View at Google Scholar · View at Scopus
- Y.-L. Lin, R.-J. Lin, K.-P. Shen et al., “Baicalein, isolated from Scutellaria baicalensis, protects against endothelin-1-induced pulmonary artery smooth muscle cell proliferation via inhibition of TRPC1 channel expression,” Journal of Ethnopharmacology, vol. 138, no. 2, pp. 373–381, 2011. View at Publisher · View at Google Scholar · View at Scopus
- J. Liang, H. Bi, and W. Ji, “Involvement of TRPA1 in ET-1-induced pain-like behavior in mice,” NeuroReport, vol. 21, no. 3, pp. 201–205, 2010. View at Publisher · View at Google Scholar · View at Scopus
- J. A. Murphy, M. L. Archibald, W. H. Baldridge, and B. C. Chauhan, “Endothelin-1-induced proliferation is reduced and Ca2+ signaling is enhanced in endothelin B-deficient optic nerve head astrocytes,” Investigative Ophthalmology and Visual Science, vol. 52, no. 10, pp. 7771–7777, 2011. View at Google Scholar · View at Scopus
- G. Bkaily, S. Choufani, L. Avedanian et al., “Nonpeptidic antagonists of ETA and ETB receptors reverse the ET-1-induced sustained increase of cytosolic and nuclear calcium in human aortic vascular smooth muscle cells,” Canadian Journal of Physiology and Pharmacology, vol. 86, no. 8, pp. 546–556, 2008. View at Publisher · View at Google Scholar · View at Scopus
- S. de Frutos, J. M. R. Diaz, C. H. Nitta, M. L. Sherpa, and L. V. Bosc, “Endothelin-1 contributes to increased NFATc3 activation by chronic hypoxia in pulmonary arteries,” American Journal of Physiology, vol. 301, no. 2, pp. C441–C450, 2011.View at Publisher · View at Google Scholar · View at Scopus
- Y. Liu, E. S. Ji, S. Xiang et al., “Exposure to cyclic intermittent hypoxia increases expression of functional NMDA receptors in the rat carotid body,” Journal of Applied Physiology, vol. 106, no. 1, pp. 259–267, 2009. View at Publisher · View at Google Scholar · View at Scopus
- S. Moyanova, L. Kortenska, and R. Mitreva, “Endothelin-1-induced cerebral ischemia: effects of ketanserin and MK-801 on limb placing in rats,” International Journal of Neuroscience, vol. 117, no. 9, pp. 1361–1381, 2007. View at Publisher ·View at Google Scholar · View at Scopus
- W. Deng, L. Baki, and C. M. Baumgarten, “Endothelin signalling regulates volume-sensitive Cl− current via NADPH oxidase and mitochondrial reactive oxygen species,” Cardiovascular Research, vol. 88, no. 1, pp. 93–100, 2010. View at Publisher · View at Google Scholar · View at Scopus
- V. C. De Giusti, M. V. Correa, M. C. Villa-Abrille et al., “The positive inotropic effect of endothelin-1 is mediated by mitochondrial reactive oxygen species,” Life Sciences, vol. 83, no. 7-8, pp. 264–271, 2008. View at Publisher · View at Google Scholar · View at Scopus
- K. F. Beck, M. G. Mohaupt, and R. B. Sterzel, “Endothelin-1 inhibits cytokine-stimulated transcription of inducible nitric oxide synthase in glomerular mesangial cells,” Kidney International, vol. 48, no. 6, pp. 1893–1899, 1995. View at Google Scholar · View at Scopus
- M. Yoshida, N. Nakanishi, X. Wang, and Y. Hattori, “Exogenous biopterins requirement for iNOS function in vascular smooth muscle cells,” Journal of Cardiovascular Pharmacology, vol. 42, no. 2, pp. 197–203, 2003. View at Publisher ·View at Google Scholar · View at Scopus
- S. Lakshminrusimha, J. A. Russell, S. Wedgwood et al., “Superoxide dismutase improves oxygenation and reduces oxidation in neonatal pulmonary hypertension,” American Journal of Respiratory and Critical Care Medicine, vol. 174, no. 12, pp. 1370–1377, 2006. View at Publisher · View at Google Scholar ·View at Scopus
- J. Herget, J. Wilhelm, J. Novotná et al., “A possible role of the oxidant tissue injury in the development of hypoxic pulmonary hypertension,” Physiological Research, vol. 49, no. 5, pp. 493–501, 2000. View at Google Scholar · View at Scopus
- N. Weissmann, R. T. Schermuly, H. A. Ghofrani et al., “Hypoxic pulmonary vasoconstriction—triggered by an increase in reactive oxygen species?” Novartis Foundation Symposium, vol. 272, pp. 196–208, 2006. View at Google Scholar ·View at Scopus
- P. Dorfmüller, M.-C. Chaumais, M. Giannakouli et al., “Increased oxidative stress and severe arterial remodeling induced by permanent high-flow challenge in experimental pulmonary hypertension,” Respiratory Research, vol. 12, Article ID 119, 2011. View at Publisher · View at Google Scholar · View at Scopus
- M. Rashid, A. Kotwani, and M. Fahim, “Long lasting phosphodiesterase 5 inhibitor, tadalafil, and superoxide dismutase mimic, tempol, protect against hypoxia-induced pulmonary hypertension in rats,” Human and Experimental Toxicology, vol. 31, no. 6, pp. 626–636, 2012. View at Google Scholar
- X. Lu, T. C. Murphy, M. S. Nanes, and C. M. Hart, “PPARγ regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-κB,” American Journal of Physiology, vol. 299, no. 4, pp. L559–L566, 2010.View at Publisher · View at Google Scholar · View at Scopus
- P. Oishi, A. Grobe, E. Benavidez et al., “Inhaled nitric oxide induced NOS inhibition and rebound pulmonary hypertension: a role for superoxide and peroxynitrite in the intact lamb,” American Journal of Physiology, vol. 290, no. 2, pp. L359–L366, 2006. View at Publisher · View at Google Scholar · View at Scopus
- E. O. Agbani, P. Coats, and R. M. Wadsworth, “Acute hypoxia stimulates intracellular peroxynitrite formation associated with pulmonary artery smooth muscle cell proliferation,” Journal of Cardiovascular Pharmacology, vol. 57, no. 5, pp. 584–588, 2011. View at Publisher · View at Google Scholar · View at Scopus
- S. Aggarwal, C. M. Gross, S. Kumar et al., “Attenuated vasodilatation in lambs with endogenous and exogenous activation of cGMP signaling: role of protein kinase G nitration,” Journal of Cellular Physiology, vol. 226, no. 12, pp. 3104–3113, 2011.View at Publisher · View at Google Scholar · View at Scopus
- E. O. Agbani, P. Coats, A. Mills, and R. M. Wadsworth, “Peroxynitrite stimulates pulmonary artery endothelial and smooth muscle cell proliferation: involvement of ERK and PKC,” Pulmonary Pharmacology and Therapeutics, vol. 24, no. 1, pp. 100–109, 2011. View at Publisher · View at Google Scholar · View at Scopus
- J. Belik, D. Stevens, J. Pan et al., “Pulmonary vascular and cardiac effects of peroxynitrite decomposition in newborn rats,” Free Radical Biology and Medicine, vol. 49, no. 8, pp. 1306–1314, 2010. View at Publisher · View at Google Scholar ·View at Scopus
- A. Masood, R. Belcastro, J. Li, C. Kantores, R. P. Jankov, and A. K. Tanswell, “A peroxynitrite decomposition catalyst prevents 60% O2-mediated rat chronic neonatal lung injury,” Free Radical Biology and Medicine, vol. 49, no. 7, pp. 1182–1191, 2010. View at Publisher · View at Google Scholar · View at Scopus
- Z. Q. Liu, B. Liu, L. Yu, X. Q. Wang, J. Wang, and H. M. Liu, “Simvastatin has beneficial effect on pulmonary artery hypertension by inhibiting NF-κB expression,” Molecular and Cellular Biochemistry, vol. 354, no. 1-2, pp. 77–82, 2011. View at Publisher · View at Google Scholar · View at Scopus
- J. Li, J. J. Li, J. G. He, J. L. Nan, Y. L. Guo, and C. M. Xiong, “Atorvastatin decreases C-reactive protein-induced inflammatory response in pulmonary artery smooth muscle cells by inhibiting nuclear factor-κb pathway,” Cardiovascular Therapeutics, vol. 28, no. 1, pp. 8–14, 2010. View at Publisher · View at Google Scholar · View at Scopus
- S. Kimura, K. Egashira, L. Chen et al., “Nanoparticle-mediated delivery of nuclear factor KB decoy into lungs ameliorates monocrotaline-induced pulmonary arterial hypertension,” Hypertension, vol. 53, no. 5, pp. 877–883, 2009. View at Publisher ·View at Google Scholar · View at Scopus
- H. Sawada, Y. Mitani, J. Maruyama et al., “A nuclear factor-κB inhibitor pyrrolidine dithiocarbamate ameliorates pulmonary hypertension in rats,” Chest, vol. 132, no. 4, pp. 1265–1274, 2007. View at Publisher · View at Google Scholar ·View at Scopus
- J. Huang, P. M. Kaminski, J. G. Edwards et al., “Pyrrolidine dithiocarbamate restores endothelial cell membrane integrity and attenuates monocrotaline-induced pulmonary artery hypertension,” American Journal of Physiology, vol. 294, no. 6, pp. L1250–L1259, 2008. View at Publisher · View at Google Scholar · View at Scopus
- M. V. Autieri, T. L. Yue, G. Z. Ferstein, and E. Ohlstein, “Antisense oligonucleotides to the p65 subunit of NF-κB inhibit human vascular smooth muscle cell adherence and proliferation and prevent neointima formation in rat carotid arteries,” Biochemical and Biophysical Research Communications, vol. 213, no. 3, pp. 827–836, 1995. View at Publisher · View at Google Scholar · View at Scopus
- B. S. Zuckerbraun, C. A. McCloskey, R. S. Mahidhara, P. K. M. Kim, B. S. Taylor, and E. Tzeng, “Overexpression of mutated IκBα inhibits vascular smooth muscle cell proliferation and intimal hyperplasia formation,” Journal of Vascular Surgery, vol. 38, no. 4, pp. 812–819, 2003. View at Publisher · View at Google Scholar · View at Scopus
- E. Soon, A. M. Holmes, C. M. Treacy et al., “Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension,” Circulation, vol. 122, no. 9, pp. 920–927, 2010. View at Publisher ·View at Google Scholar · View at Scopus
- M. Li, S. R. Riddle, M. G. Frid, et al., “Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension,” Journal of Immunology, vol. 187, no. 5, pp. 2711–2722, 2011. View at Publisher · View at Google Scholar · View at Scopus
- T. M. Yu, Y. H. Chen, J. Y. Hsu et al., “Systemic inflammation is associated with pulmonary hypertension in patients undergoing haemodialysis,” Nephrology Dialysis Transplantation, vol. 24, no. 6, pp. 1946–1951, 2009. View at Publisher ·View at Google Scholar · View at Scopus
- D. D. Sin and S. F. P. Man, “Is systemic inflammation responsible for pulmonary hypertension in COPD?” Chest, vol. 130, no. 2, pp. 310–312, 2006. View at Publisher · View at Google Scholar · View at Scopus
- K. R. Hamal, R. F. Wideman, N. B. Anthony, and G. F. Erf, “Differential gene expression of proinflammatory chemokines and cytokines in lungs of ascites-resistant and -susceptible broiler chickens following intravenous cellulose microparticle injection,” Veterinary Immunology and Immunopathology, vol. 133, no. 2–4, pp. 250–255, 2010. View at Publisher · View at Google Scholar · View at Scopus
- N. F. Voelkel, R. M. Tuder, J. Bridges, and W. P. Arend, “Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline,” American Journal of Respiratory Cell and Molecular Biology, vol. 11, no. 6, pp. 664–675, 1994. View at Google Scholar · View at Scopus
- A. A. Wanderer, “Rationale for IL-1β targeted therapy for ischemia-reperfusion induced pulmonary and other complications in sickle cell disease,” Journal of Pediatric Hematology/Oncology, vol. 31, no. 8, pp. 537–538, 2009. View at Publisher · View at Google Scholar · View at Scopus
- G. N. Kalambokis, A. Mouzaki, M. Rodi, K. Pappas, P. Korantzopoulos, and E. V. Tsianos, “Serum interleukin 6 levels and cirrhosis-associated pulmonary hypertension,” Angiology, vol. 62, no. 4, pp. 344–345, 2011. View at Publisher ·View at Google Scholar · View at Scopus
- A. Chaouat, L. Savale, C. Chouaid et al., “Role for interleukin-6 in COPD-related pulmonary hypertension,” Chest, vol. 136, no. 3, pp. 678–687, 2009. View at Publisher · View at Google Scholar · View at Scopus
- Y. Furuya, T. Satoh, and M. Kuwana, “Interleukin-6 as a potential therapeutic target for pulmonary arterial hypertension,” International Journal of Rheumatology, vol. 2010, Article ID 720305, 2010. View at Publisher · View at Google Scholar · View at Scopus
- M. K. Steiner, O. L. Syrkina, N. Kolliputi, E. J. Mark, C. A. Hales, and A. B. Waxman, “Interleukin-6 overexpression induces pulmonary hypertension,”Circulation Research, vol. 104, no. 2, pp. 236–244, 2009. View at Publisher · View at Google Scholar · View at Scopus
- M. Seimetz, N. Parajuli, A. Pichl et al., “Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice,” Cell, vol. 147, no. 2, pp. 293–305, 2011. View at Publisher · View at Google Scholar · View at Scopus
- J. Moral-Sanz, C. Menendez, L. Moreno, E. Moreno, A. Cogolludo, and F. Perez-Vizcaino, “Pulmonary arterial dysfunction in insulin resistant obese Zucker rats,”Respiratory Research, vol. 12, article 51, 2011. View at Publisher · View at Google Scholar · View at Scopus
- T. P. Shanley, B. Zhao, D. R. Macariola, A. Denenberg, A. L. Salzman, and P. A. Ward, “Role of nitric oxide in acute lung inflammation: lessons learned from the inducible nitric oxide synthase knockout mouse,” Critical Care Medicine, vol. 30, no. 9, pp. 1960–1968, 2002. View at Google Scholar · View at Scopus
- G. Sutendra, P. Dromparis, P. Wright et al., “The role of nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension,” Science Translational Medicine, vol. 3, no. 88, Article ID 88ra55, 2011. View at Publisher ·View at Google Scholar · View at Scopus
- K. N. Farrow, S. Wedgwood, K. J. Lee et al., “Mitochondrial oxidant stress increases PDE5 activity in persistent pulmonary hypertension of the newborn,”Respiratory Physiology and Neurobiology, vol. 174, no. 3, pp. 272–281, 2010. View at Publisher · View at Google Scholar · View at Scopus
- J. Rehman and S. L. Archer, “A proposed mitochondrial-metabolic mechanism for initiation and maintenance of pulmonary arterial hypertension in fawn-hooded rats: the warburg model of pulmonary arterial hypertension,” Advances in Experimental Medicine and Biology, vol. 661, pp. 171–185, 2010. View at Publisher· View at Google Scholar · View at Scopus
- R. Belostotsky, E. Ben-Shalom, C. Rinat et al., “Mutations in the mitochondrial Seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome,” American Journal of Human Genetics, vol. 88, no. 2, pp. 193–200, 2011. View at Publisher · View at Google Scholar · View at Scopus
- S. L. Archer, M. Gomberg-Maitland, M. L. Maitland, S. Rich, J. G. N. Garcia, and E. K. Weir, “Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1α-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer,” American Journal of Physiology, vol. 294, no. 2, pp. H570–H578, 2008. View at Publisher · View at Google Scholar · View at Scopus
- N. Weissmann, N. Ebert, M. Ahrens et al., “Effects of mitochondrial inhibitors and uncouplers on hypoxic vasoconstriction in rabbit lungs,” American Journal of Respiratory Cell and Molecular Biology, vol. 29, no. 6, pp. 721–732, 2003. View at Publisher · View at Google Scholar · View at Scopus
- K. Shimoda, K. Murakami, P. Enkhbaatar et al., “Effect of poly(ADP ribose) synthetase inhibition on burn and smoke inhalation injury in sheep,” American Journal of Physiology, vol. 285, no. 1, pp. L240–L249, 2003. View at Google Scholar· View at Scopus
- A. Tasatargil, G. Sadan, and E. Karasu, “Homocysteine-induced changes in vascular reactivity of guinea-pig pulmonary arteries: role of the oxidative stress and poly (ADP-ribose) polymerase activation,” Pulmonary Pharmacology and Therapeutics, vol. 20, no. 3, pp. 265–272, 2007. View at Publisher · View at Google Scholar · View at Scopus
- Y. Yu, S. H. Keller, C. V. Remillard et al., “A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension,” Circulation, vol. 119, no. 17, pp. 2313–2322, 2009. View at Publisher · View at Google Scholar · View at Scopus
- M. I. Townsley, J. A. King, and D. F. Alvarez, “Ca2+ channels and pulmonary endothelial permeability: Insights from study of intact lung and chronic pulmonary hypertension,” Microcirculation, vol. 13, no. 8, pp. 725–739, 2006. View at Publisher · View at Google Scholar · View at Scopus
- A. L. Firth, C. V. Remillard, and J. X. J. Yuan, “TRP channels in hypertension,”Biochimica et Biophysica Acta, vol. 1772, no. 8, pp. 895–906, 2007. View at Publisher · View at Google Scholar · View at Scopus
- K. R. Zhou and Y. L. Lai, “Capsaicin pretreatment attenuates monocrotaline-induced ventilatory dysfunction and pulmonary hypertension,” Journal of Applied Physiology, vol. 75, no. 6, pp. 2781–2788, 1993. View at Google Scholar · View at Scopus
- Y. L. Lai, C. F. Chen, C. T. Chien, H. L. Shiao, A. A. Thacker, and H. Q. Zhang, “Capsaicin pretreatment attenuates chronic hypoxic pulmonary hypertension,”Respiration Physiology, vol. 99, no. 2, pp. 283–289, 1995. View at Publisher · View at Google Scholar · View at Scopus
- N. J. Katzman and Y. L. Lai, “Capsaicin pre- and post-treatment on rat monocrotaline pneumotoxicity,” Chinese Journal of Physiology, vol. 43, no. 4, pp. 171–178, 2000. View at Google Scholar · View at Scopus
- R. Goyal, D. G. Papamatheakis, M. Loftin et al., “Long-term maternal hypoxia: the role of extracellular Ca2+ entry during serotonin-mediated contractility in fetal ovine pulmonary arteries,” Reproductive Sciences, vol. 18, no. 10, pp. 948–962, 2011. View at Publisher · View at Google Scholar · View at Scopus
- A. R. Tonelli, H. Alnuaimat, and K. Mubarak, “Pulmonary vasodilator testing and use of calcium channel blockers in pulmonary arterial hypertension,” Respiratory Medicine, vol. 104, no. 4, pp. 481–496, 2010. View at Publisher · View at Google Scholar · View at Scopus
- D. Montani, L. Savale, D. Natali et al., “Long-term response to calcium-channel blockers in non-idiopathic pulmonary arterial hypertension,” European Heart Journal, vol. 31, no. 15, pp. 1898–1907, 2010. View at Publisher · View at Google Scholar · View at Scopus
- Y. Yang, M. Gao, Y. Guo, and J. Qiao, “Calcium antagonists, diltiazem and nifedipine, protect broilers against low temperature-induced pulmonary hypertension and pulmonary vascular remodeling,” Animal Science Journal, vol. 81, no. 4, pp. 494–500, 2010. View at Publisher · View at Google Scholar · View at Scopus
- Y. Yang, J. Qiao, H. Wang et al., “Calcium antagonist verapamil prevented pulmonary arterial hypertension in broilers with ascites by arresting pulmonary vascular remodeling,” European Journal of Pharmacology, vol. 561, no. 1–3, pp. 137–143, 2007. View at Publisher · View at Google Scholar · View at Scopus
- W. Ma, W. Han, P. A. Greer et al., “Calpain mediates pulmonary vascular remodeling in rodent models of pulmonary hypertension, and its inhibition attenuates pathologic features of disease,” Journal of Clinical Investigation, vol. 121, no. 11, pp. 4548–4566, 2011. View at Publisher · View at Google Scholar · View at Scopus
- A. Malinovschi, D. Henrohn, A. Eriksson, J. O. Lundberg, K. Alving, and G. Wikström, “Increased plasma and salivary nitrite and decreased bronchial contribution to exhaled NO in pulmonary arterial hypertension,” European Journal of Clinical Investigation, vol. 41, no. 8, pp. 889–897, 2011. View at Publisher · View at Google Scholar · View at Scopus
- R. E. Girgis, H. C. Champion, G. B. Diette, R. A. Johns, S. Permutt, and J. T. Sylvester, “Decreased exhaled nitric oxide in pulmonary arterial hypertension: response to Bosentan therapy,” American Journal of Respiratory and Critical Care Medicine, vol. 172, no. 3, pp. 352–357, 2005. View at Publisher · View at Google Scholar · View at Scopus
- F. T. Kaneko, A. C. Arroliga, R. A. Dweik et al., “Biochemical reaction products of nitric oxide as quantitative markers of primary pulmonary hypertension,”American Journal of Respiratory and Critical Care Medicine, vol. 158, no. 3, pp. 917–923, 1998. View at Google Scholar · View at Scopus
- G. Rolla, P. Colagrande, E. Scappaticci et al., “Exhaled nitric oxide in systemic sclerosis: relationships with lung involvement and pulmonary hypertension,”Journal of Rheumatology, vol. 27, no. 7, pp. 1693–1698, 2000. View at Google Scholar · View at Scopus
- M. S. Riley, J. Pórszász, J. Miranda, M. P. K. J. Engelen, B. Brundage, and K. Wasserman, “Exhaled nitric oxide during exercise in primary pulmonary hypertension and pulmonary fibrosis,” Chest, vol. 111, no. 1, pp. 44–50, 1997. View at Google Scholar · View at Scopus
- S. L. Archer, K. Djaballah, M. Humbert et al., “Nitric oxide deficiency in fenfluramine- and dexfenfluramine-induced pulmonary hypertension,” American Journal of Respiratory and Critical Care Medicine, vol. 158, no. 4, pp. 1061–1067, 1998. View at Google Scholar · View at Scopus
- Z. Gölbaş, S. Dinçer, H. Bayol et al., “Increased nitric oxide in exhaled air in patients with rheumatic heart disease,” European Journal of Heart Failure, vol. 3, no. 1, pp. 27–32, 2001. View at Publisher · View at Google Scholar · View at Scopus
- C. R. Forrest, C. Y. Pang, A. G. Zhong, and M. L. Kreidstein, “Efficacy of intravenous infusion of prostacyclin (PGI2) or prostaglandin E1 (PGE1) in augmentation of skin flap blood flow and viability in the pig,” Prostaglandins, vol. 41, no. 6, pp. 537–558, 1991. View at Publisher · View at Google Scholar · View at Scopus
- M. Özkan, R. A. Dweik, D. Laskowski, A. C. Arroliga, and S. C. Erzurum, “High levels of nitric oxide in individuals with pulmonary hypertension receiving epoprostenol therapy,” Lung, vol. 179, no. 4, pp. 233–243, 2001. View at Publisher ·View at Google Scholar · View at Scopus
- B. Weinberger, L. Fakhrzadeh, D. E. Heck, J. D. Laskin, C. R. Gardner, and D. L. Laskin, “Inhaled nitric oxide primes lung macrophages to produce reactive oxygen and nitrogen intermediates,” American Journal of Respiratory and Critical Care Medicine, vol. 158, no. 3, pp. 931–938, 1998. View at Google Scholar · View at Scopus
- I. M. Robbins, A. R. Hemnes, J. Simon Gibbs et al., “Safety of sapropterin dihydrochloride (6r-bh4) in patients with pulmonary hypertension,” Experimental Lung Research, vol. 37, no. 1, pp. 26–34, 2011. View at Publisher · View at Google Scholar · View at Scopus
- R.-J. Teng, J. Du, H. Xu et al., “Sepiapterin improves angiogenesis of pulmonary artery endothelial cells with in utero pulmonary hypertension by recoupling endothelial nitric oxide synthase,” American Journal of Physiology, vol. 301, no. 3, pp. L334–L345, 2011. View at Publisher · View at Google Scholar · View at Scopus
- M. Umehara, A. Yamaguchi, S. Itakura et al., “Repeated Waon therapy improves pulmonary hypertension during exercise in patients with severe chronic obstructive pulmonary disease,” Journal of Cardiology, vol. 51, no. 2, pp. 106–113, 2008. View at Publisher · View at Google Scholar · View at Scopus
- M. L. Pall, “Do sauna therapy and exercise act by raising the availability of tetrahydrobiopterin?” Medical Hypotheses, vol. 73, no. 4, pp. 610–613, 2009. View at Publisher · View at Google Scholar · View at Scopus
- F. Raimondi, F. Migliaro, L. Capasso et al., “Intravenous magnesium sulphate vs. inhaled nitric oxide for moderate, persistent pulmonary hypertension of the newborn. A multicentre, retrospective study,” Journal of Tropical Pediatrics, vol. 54, no. 3, pp. 196–199, 2008. View at Publisher · View at Google Scholar · View at Scopus
- S. H. Daffa and W. A. Milaat, “Role of magnesium sulphate in treatment of severe persistent pulmonary hypertension of the neoborn,” Saudi Medical Journal, vol. 23, no. 10, pp. 1266–1269, 2002. View at Google Scholar · View at Scopus
- S. Chandran, E. Haque, H. T. Wickramasinghe, and Z. Wint, “Use of magnesium sulphate in severe persistent pulmonary hypertension of the newborn,” Journal of Tropical Pediatrics, vol. 50, no. 4, pp. 219–223, 2004. View at Publisher · View at Google Scholar · View at Scopus
- J. F. Tolsa, J. Cotting, N. Sekarski, M. Payot, J. L. Micheli, and A. Calame, “Magnesium sulphate as an alternative and safe treatment for severe persistent pulmonary hypertension of the newborn,” Archives of Disease in Childhood, vol. 72, no. 3, pp. F184–F187, 1995. View at Google Scholar · View at Scopus
- S. Uslu, S. Kumtepe, A. Bulbul, S. Comert, F. Bolat, and A. Nuhoglu, “A comparison of magnesium sulphate and sildenafil in the treatment of the newborns with persistent pulmonary hypertension: a randomized controlled trial,” Journal of Tropical Pediatrics, vol. 57, no. 4, pp. 245–250, 2011. View at Publisher · View at Google Scholar · View at Scopus
- N. Y. Boo, J. Rohana, S. C. Yong, A. Z. Bilkis, and F. Yong-Junina, “Inhaled nitric oxide and intravenous magnesium sulphate for the treatment of persistent pulmonary hypertension of the newborn,” Singapore Medical Journal, vol. 51, no. 2, pp. 144–150, 2010. View at Google Scholar · View at Scopus
- A. Csiszar, N. Labinskyy, A. Podlutsky et al., “Vasoprotective effects of resveratrol and SIRT1: attenuation of cigarette smoke-induced oxidative stress and proinflammatory phenotypic alterations,” American Journal of Physiology, vol. 294, no. 6, pp. H2721–H2735, 2008. View at Publisher · View at Google Scholar · View at Scopus
- A. Csiszar, N. Labinskyy, S. Olson et al., “Resveratrol prevents monocrotaline-induced pulmonary hypertension in rats,” Hypertension, vol. 54, no. 3, pp. 668–675, 2009. View at Publisher · View at Google Scholar · View at Scopus
- Z. Ungvari, N. Labinskyy, P. Mukhopadhyay et al., “Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells,” American Journal of Physiology, vol. 297, no. 5, pp. H1876–H1881, 2009. View at Publisher ·View at Google Scholar · View at Scopus
- A. Biala, E. Tauriainen, A. Siltanen et al., “Resveratrol induces mitochondrial biogenesis and ameliorates Ang II-induced cardiac remodeling in transgenic rats harboring human renin and angiotensinogen genes,” Blood Pressure, vol. 19, no. 3, pp. 196–205, 2010. View at Publisher · View at Google Scholar · View at Scopus
- N. Xia, A. Daiber, A. Habermeier et al., “Resveratrol reverses endothelial nitric-oxide synthase uncoupling in apolipoprotein E knockout mice,” Journal of Pharmacology and Experimental Therapeutics, vol. 335, no. 1, pp. 149–154, 2010.View at Publisher · View at Google Scholar · View at Scopus
- Z. B. Gao, X. Q. Chen, and G. Y. Hu, “Inhibition of excitatory synaptic transmission by trans-resveratrol in rat hippocampus,” Brain Research, vol. 1111, no. 1, pp. 41–47, 2006. View at Publisher · View at Google Scholar · View at Scopus
- H. Zhang, G. P. Schools, T. Lei, W. Wang, H. K. Kimelberg, and M. Zhou, “Resveratrol attenuates early pyramidal neuron excitability impairment and death in acute rat hippocampal slices caused by oxygen-glucose deprivation,”Experimental Neurology, vol. 212, no. 1, pp. 44–52, 2008. View at Publisher · View at Google Scholar · View at Scopus
- A. P. Raval, H. W. Lin, K. R. Dave et al., “Resveratrol and ischemic preconditioning in the brain,” Current Medicinal Chemistry, vol. 15, no. 15, pp. 1545–1551, 2008.View at Publisher · View at Google Scholar · View at Scopus
- A. Quincozes-Santos and C. Gottfried, “Resveratrol modulates astroglial functions: neuroprotective hypothesis,” Annals of the New York Academy of Sciences, vol. 1215, no. 1, pp. 72–78, 2011. View at Publisher · View at Google Scholar · View at Scopus
- L. G. Chicoine, J. A. Stewart Jr., and P. A. Lucchesi, “Is resveratrol the magic bullet for pulmonary hypertension?” Hypertension, vol. 54, no. 3, pp. 473–474, 2009.View at Publisher · View at Google Scholar · View at Scopus
- M. C. Zillikens, J. B. J. van Meurs, E. J. G. Sijbrands et al., “SIRT1 genetic variation and mortality in type 2 diabetes: interaction with smoking and dietary niacin,” Free Radical Biology and Medicine, vol. 46, no. 6, pp. 836–841, 2009. View at Publisher ·View at Google Scholar · View at Scopus
 RSS Feed
RSS Feed
